 ADC drugs utilize monoclonal antibodies to specifically deliver cytotoxic drugs to tumor cells, characterized by precise targeting and specific killing of tumors, thereby limiting the exposure of cytotoxic drugs to normal cells or tissues. The main clinical toxicities of ADC drugs include gastrointestinal, hematological, hepatic, neurological, and ocular toxicities, which are usually dose-limiting[2]. Depending on whether they specifically bind to the action target, the treatment-related toxicities of ADC drugs can be classified into on-target toxicity (target-dependent) and off-target toxicity (non-target dependent)[2]. Understanding the mechanisms of target-dependent and/or non-target-dependent cytotoxicity induced by ADC drugs is crucial for future development strategies of ADC drugs. T-DM1 is the first ADC drug introduced in the field of solid tumors, with nearly 10 years of clinical application, and the exploration of its adverse reaction mechanisms is relatively mature. This article takes T-DM1 as an example to primarily discuss the mechanisms of T-DM1-induced thrombocytopenia and hepatotoxicity, and thus expands on the thoughts regarding off-target toxicity of ADC drugs..
ADC drugs utilize monoclonal antibodies to specifically deliver cytotoxic drugs to tumor cells, characterized by precise targeting and specific killing of tumors, thereby limiting the exposure of cytotoxic drugs to normal cells or tissues. The main clinical toxicities of ADC drugs include gastrointestinal, hematological, hepatic, neurological, and ocular toxicities, which are usually dose-limiting[2]. Depending on whether they specifically bind to the action target, the treatment-related toxicities of ADC drugs can be classified into on-target toxicity (target-dependent) and off-target toxicity (non-target dependent)[2]. Understanding the mechanisms of target-dependent and/or non-target-dependent cytotoxicity induced by ADC drugs is crucial for future development strategies of ADC drugs. T-DM1 is the first ADC drug introduced in the field of solid tumors, with nearly 10 years of clinical application, and the exploration of its adverse reaction mechanisms is relatively mature. This article takes T-DM1 as an example to primarily discuss the mechanisms of T-DM1-induced thrombocytopenia and hepatotoxicity, and thus expands on the thoughts regarding off-target toxicity of ADC drugs..
T-DM1 Toxicity and Off-Target Mechanisms Overview
T-DM1 is formed by coupling the cytotoxic drug DM1 through an indissoluble thioether linker with the humanized monoclonal antibody trastuzumab. After binding to HER2, T-DM1 is internalized into tumor cells and degraded in lysosomes, forming the Lys-MCC-DM1 complex as the effector molecule, targeting tubulin to induce cell death. Over the past nearly 10 years, T-DM1 has served as a second-line standard treatment for HER2-positive advanced breast cancer and a reinforced adjuvant therapy option for HER2-positive early breast cancer patients with residual invasive disease after neoadjuvant therapy. Thrombocytopenia, elevated liver transaminases, and peripheral neuropathy are common adverse reactions of T-DM1[3]. The major dose-limiting toxicities observed in clinical trials for T-DM1 are thrombocytopenia and elevated liver transaminases[2].
Off-target effects are currently the main cause of dose-limiting toxicities in the clinical development of ADC drugs[2]. Possible mechanisms include ① linker-drug instability leading to premature release of the drug in circulation; ② bystander effects; ③ normal cells uptake/transport the intact ADC drugs through receptor-dependent and non-receptor-dependent (non-specific endocytosis) mechanisms[4]. Although the non-target-dependent uptake of ADC drugs by normal cells may be one of the main reasons for off-target toxicity, this mechanism is relatively under-researched in ADC drug-related toxicities[2].
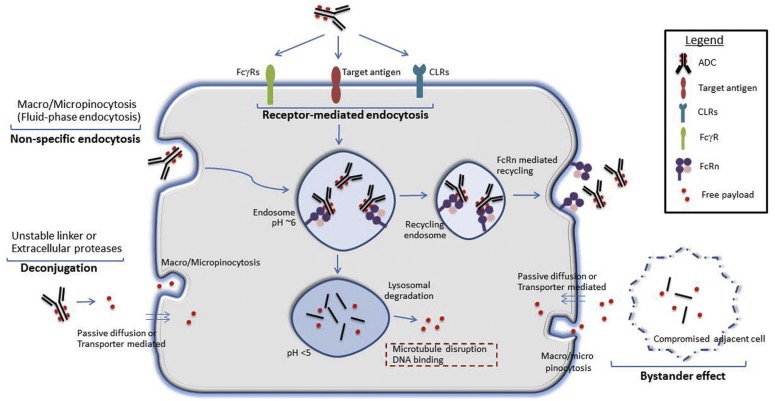
Figure 1 Potential mechanisms of normal cell uptake of ADC drugs or free payloads. ① Target antigens may be expressed on normal cells, facilitating the target-dependent uptake of ADC drugs. ② Receptors binding to the Fc region of IgG antibodies, such as Fcγ receptors (FcγR), neonatal Fc receptor (FcRn), and C-type lectin receptors (CLR), may also facilitate non-target-dependent endocytosis/transport of ADC drugs in normal cells. ③ Non-specific endocytosis mechanisms, such as macropinocytosis or micropinocytosis, may also lead to the entry of intact ADC drugs or free payloads (due to linker-drug instability or extracellular protease activity causing the payload to be released outside the cell) into cells. ④ Free payloads may also enter normal cells through other mechanisms, such as passive diffusion (if membrane-permeable), non-specific endocytosis, or uptake mediated by specific transport proteins (if they are substrates of membrane transport proteins). ⑤ Antigen-positive target cells may also mediate toxicity by releasing the payload into the local environment, which is subsequently taken up by antigen-negative normal cells through passive diffusion, transporter-mediated uptake, or other non-specific endocytosis mechanisms, similar to the bystander effect[4].
It is known that antibodies can be taken up by normal cells in a non-target-dependent manner through various mechanisms, including Fc receptor-mediated endocytosis pathways (e.g., mannose receptors, FcRn, and FcγR receptors), non-specific endocytosis, and/or uptake of ADC drug metabolites. Theoretically, non-target-dependent uptake could lead to the intracellular transport of ADC drugs via endosomal or lysosomal pathways, followed by the release of cytotoxic payloads in normal cells. However, there is very little data evaluating the impact of non-target-dependent uptake on the overall off-target toxicity profile of ADC drugs. The most extensive evidence supporting this hypothesis comes from studies exploring the mechanism of T-DM1-mediated thrombocytopenia[2].
Moreover, considering that the maximum tolerated dose (MTD) range for most ADC drugs that bind to microtubule inhibitors is 2-5 mg/kg regardless of target protein expression, it is essential to gain a more in-depth understanding of the potential mechanisms of off-target toxicity to improve the therapeutic index of next-generation ADC drugs[2].
Mechanisms of T-DM1 Induced Thrombocytopenia
Hematological toxicity is the most common non-target-dependent dose-limiting toxicity of ADC drugs using auristatins (e.g., MMAE, MMAF), calicheamicins, and maytansinoids (e.g., DM1) as “payload” molecules[4].
Researchers Uppal et al.[4,5] studied the mechanisms of T-DM1-mediated thrombocytopenia and supported the critical role of FcγR in ADC drug treatment-related hematological toxicity. Previous studies have confirmed that HER2 expression is not found on the surface of circulating platelets or megakaryocytes (MKs) differentiated from bone marrow, so it is expected that there is no target-dependent uptake in these cells. Uppal et al. utilized in vitro experiments and a novel human bone marrow hematopoietic stem/progenitor cell differentiation model, which not only ruled out the direct effect of ADC on mature platelets but also revealed that FcγRIIa-mediated internalization of T-DM1 in megakaryocytes (in the bone marrow) and the resulting cytotoxicity (cytoskeletal disruption) is the mechanism for reduced platelet production. Using FcγRII blocking antibodies (anti-CD32 antibodies) or using Fcγ mutants of T-DM1 that cannot bind to FcγR (T-DM1-DANA carrying D265A and N297A mutations) significantly reduced T-DM1 internalization in megakaryocytes. Additionally, T-DM1 and control ADC drugs containing DM1 were observed to affect megakaryocytes, while trastuzumab, although also internalized by megakaryocytes, did not have adverse effects, indicating that the damage caused by T-DM1 to megakaryocytes leading to thrombocytopenia is mediated by the payload DM1, but requires the interaction between the Fc domain on the T-DM1 antibody and FcγRIIa for internalization. Overall, the researchers concluded that FcγRIIa (at least in part) contributes to the mechanism of T-DM1 binding and internalization that mediates impaired megakaryocyte differentiation, ultimately leading to thrombocytopenia. However, the study did not support the co-localization of internalized T-DM1 with lysosomal marker LAMP1 in megakaryocytes, so it remains unclear how the ADC/FcγRIIa complex internalizes and how the effector molecule Lys-MCC-DM1 is released into the microtubules of megakaryocytes[3].
In contrast to the results of this study, researchers Thon et al.[6] believe that T-DM1-induced thrombocytopenia occurs through mechanisms independent of HER2 or Fc receptors, based on Thon et al.’s co-culture of mouse fetal liver cell cultures and mature megakaryocytes with T-DM1, 5B6-DM1 (non-specific humanized antibody conjugate), trastuzumab (the antibody alone), or solvent controls. This is based on the fact that megakaryocytes/platelets do not express HER2 receptors, and that mice do not contain human IgG’s FcRIIA. Although trastuzumab is a humanized monoclonal antibody that directly targets the extracellular domain of HER2, the observed effects of 5B6-DM1 in mouse hematopoietic stem and megakaryocyte cultures are similar to those of T-DM1, indicating that the uptake of T-DM1 does not depend on HER2 and Fc RIIA. Furthermore, this study also showed that T-DM1 can disrupt the microtubule organization in megakaryocytes and platelets. At the same time, Thon also supports Uppal’s view that trastuzumab monotherapy has no effect on megakaryocyte differentiation or platelet production, and that thrombocytopenia originates from microtubule inhibition caused by the payload DM1.
Moreover, although Uppal et al. confirmed that blocking the interaction between T-DM1 and FcγRIIa significantly reduces T-DM1 internalization, it does not completely block megakaryocyte uptake of T-DM1 or cytotoxicity, suggesting that other mechanisms (such as non-specific endocytosis) may also lead to thrombocytopenia. Researchers Zhao et al.[7] published a report that supports this view well; this report studied AGS-16C3F [an ADC targeting ENPP3 (ectonucleotide pyrophosphatase/phosphodiesterase family member-3) coupled with MMAF through an indissoluble linker)] and T-DM1, using a similar in vitro megakaryocyte differentiation platform to induce thrombocytopenia. AGS-16C3F, similar to T-DM1, has no direct effect on mature platelets and does not detect ENPP3 (the target antigen) expression on circulating platelets or their precursor megakaryocytes. However, contrary to Uppal, Zhao et al. believe that the non-target-dependent macropinocytosis and its differentiation inhibition effect of ADC drugs in megakaryocytes play a role in ADC drug-induced thrombocytopenia[4,7].
ADC drugs may also induce thrombocytopenia through other mechanisms, such as peripheral destruction or increased clearance/isolation of circulating platelets. The metabolic cycle of human platelets is normally 9-11 days, after which they are cleared through various mechanisms, including desialylation, exposing potential galactose residues (recognized by Ashwell-Morrell receptors) for clearance by hepatocytes and immune-mediated (antibody or T cell-dependent) mechanisms. Understanding the changes in platelet dynamics after ADC drug treatment may help distinguish whether thrombocytopenia is due to reduced production or peripheral destruction or clearance. If the platelet count drops rapidly (acute) (< 5-7 days), it indicates accelerated destruction of platelets in peripheral blood or isolation at distal sites of damage, rather than reduced bone marrow production. For example, the thrombocytopenia induced by calicheamicin-conjugated ADC drugs in crabs was shown to be unrelated to FcγR-mediated or macropinocytosis-mediated uptake by megakaryocytes. In contrast, this hepatotoxicity primarily in liver sinusoidal endothelial cells (LSEC) is related to platelet isolation in the liver sinusoidal space, leading to the occurrence of thrombocytopenia[4].
Mechanisms of T-DM1 Induced Hepatotoxicity
HER2-Dependent Pathway
Hepatotoxicity is one of the black box warnings for T-DM1 treatment, which may lead to severe elevations in liver enzymes, serum alanine aminotransferase (ALT), and aspartate aminotransferase (AST)[3]. To explore the molecular mechanisms of T-DM1-induced hepatotoxicity, Endo et al.[8] established in vitro and in vivo models, such as immortalized human liver cells, compared to HER2-positive breast cancer cell lines sensitive to T-DM1 treatment, such as SKBR-3 and BT-474, which have low HER2 expression levels. The results indicated that T-DM1 may cause hepatotoxicity through HER2-dependent uptake pathways. Moreover, the researchers also found that T-DM1-mediated liver cell injury could be further enhanced by the pro-inflammatory cytokine TNF-α, due to T-DM1-induced rupture of the outer mitochondrial membrane, triggering mitochondrial-dependent apoptosis[9].
Target-dependent hepatotoxicity induced by ADC drugs is also very typical in gemtuzumab ozogamicin (GO, an ADC drug conjugated with anti-CD33 monoclonal antibody and calicheamicin derivative), which was approved by the FDA in 2000 for the treatment of acute myeloid leukemia (AML) and withdrawn from the market in 2010 due to post-marketing studies not showing improved survival compared to chemotherapy alone and safety issues, including significant hepatotoxicity. Maniecki et al. demonstrated that the CD33 receptor is widely distributed in liver tissues and hepatocytes. Thus, it is believed that the specific targeting of liver cells expressing CD33 by GO is related to hepatotoxicity[3].
CKAP5-Dependent Pathway
In addition to HER2-dependent hepatotoxicity induced by T-DM1, Endo et al.[9] also noted that DM1 conjugated to non-targeting IgG could also cause dose-dependent cell growth inhibition in vitro. Endo et al. hypothesized that HER2-independent uptake mechanisms may also be involved in T-DM1-induced hepatotoxicity. To identify new target molecules that may participate in T-DM1-induced hepatotoxicity, T-DM1 was used as bait and co-cultured with human and mouse liver cells in cell culture dishes for immunoprecipitation analysis targeting cell surface molecules that could bind to T-DM1. The study identified a protein with a molecular weight of 230 kDa that specifically binds to T-DM1 but does not bind to trastuzumab or control IgG
[9]. This protein was identified by mass spectrometry as cytoskeleton-associated protein 5 (CKAP5, also known as ch-TOG or XMAP215), a member of the XMAP215/Dis 1 family, which plays a key role in regulating microtubule polymerization by binding to tubulin. Regarding the tissue distribution of CKAP5, based on the Human Protein Atlas, CKAP5 is widely expressed in various human tissues[3].
Endo et al. further confirmed that the binding of CKAP5 to T-DM1 is mediated by the DM1 portion of T-DM1, and demonstrated that the interaction between DM1 and CKAP5 occurs at the cell surface, unrelated to the antibody portion of T-DM1, trastuzumab. Data showed that after binding to CKAP5 on the cell surface, T-DM1 begins to damage the cell membrane, leading to calcium influx into liver cells, resulting in microtubule disruption and cell apoptosis[9]. In this study, MMAE-conjugated vedotin did not bind to CKAP5 and did not cause cell membrane damage or hepatocyte apoptosis[9].
Endo et al. identified a new mechanism of non-target (HER2) dependent cytotoxicity mediated by ADC drugs (T-DM1) in normal cells/tissues with low HER2 expression levels. Additionally, Endo et al. found that this new mechanism may also be related to hepatotoxicity induced by other DM1 and DM4 conjugated ADC drugs, as non-specific ADC drug molecules (α-CD22-DM1 and α-gD-DM4) were observed to bind to CKAP5 and induce microtubule disruption in hepatocytes[9]. These data further suggest that CKAP5 can be considered a cell target for off-target toxicity mediated by maytansinoid (DM1 or DM4) conjugated ADC drugs, warranting further preclinical and clinical research. Figure 1 summarizes the mechanisms of hepatotoxicity induced by T-DM1 through HER2 and CKAP5-dependent pathways.
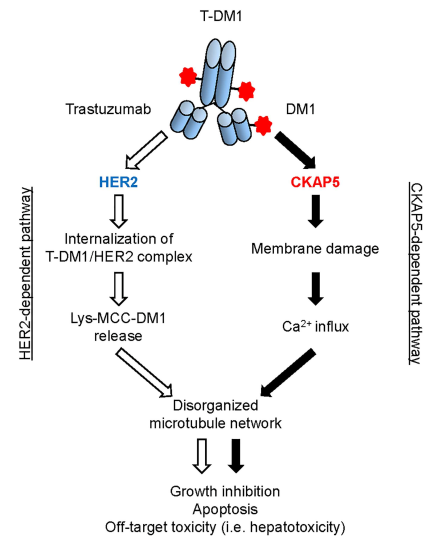
Figure 1 The mechanisms of hepatotoxicity induced by T-DM1 through HER2 and CKAP5-dependent pathways. ① HER2-dependent pathway: After T-DM1 interacts with HER2 expressed on tumor cell surfaces, the T-DM1/HER2 complex is internalized, followed by degradation in lysosomes and release of Lys-MCC-DM1 to the microtubules of target cells, leading to cell apoptosis. The HER2-dependent pathway is the main mechanism by which T-DM1 induces hepatotoxicity in HER2-positive tumor cells. ② CKAP5-dependent pathway: After T-DM1 (DM1) specifically binds to CKAP5 on the surface of hepatocytes, T-DM1 begins to damage the cell membrane, leading to calcium influx into hepatocytes. The increase in calcium concentration in the cytoplasm of hepatocytes disrupts the microtubule network, resulting in cell growth inhibition, apoptosis, and hepatotoxicity[3].
Summary and Insights
Maytansinoid-conjugated ADC drugs occupy an important proportion among the ADC drugs currently under clinical research. The side effects observed in clinical studies of these ADC drugs seem to be related to the payloads DM1 and DM4. For example, gastrointestinal reactions, thrombocytopenia, and neutropenia are associated with DM1-conjugated ADC drugs, while ocular toxicity is one of the most common adverse reactions of DM4-conjugated ADC drugs, and hepatotoxicity has been observed in both DM1 and DM4 conjugated ADC drugs.
A meta-analysis of 660 published ADC drug-related articles from 2000 to January 2014 showed significant differences in several key toxicities between DM1 and DM4 conjugated ADC drugs. Furthermore, Endo et al. also observed differences in binding activity to CKAP5 between DM1 and DM4 conjugated ADC drugs, which may be related to the different toxicity profiles detected in their respective ADC drugs.
Currently, the understanding of the mechanisms of off-target toxicity related to ADC drugs remains unclear, but the research by Endo et al. has promoted the understanding of the mechanisms of off-target toxicity induced by T-DM1. Additionally, related studies suggest that future design of ADC drugs, especially maytansinoid-conjugated ADC drugs, may benefit from strategies to conceal the payload portion of ADC drugs before intracellular uptake to reduce non-target-dependent but payload-related adverse reactions. Moreover, it is necessary to conduct more research work to explore the relevant mechanisms of off-target toxicity induced by ADC drugs in the future, laying a solid foundation for better management of ADC drug treatment-related toxicities.
References
[1] Masters JC, Nickens DJ, Xuan D, et al. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018 Feb;36(1):121-135.
[2] Hinrichs MJ, Dixit R. Antibody Drug Conjugates: Nonclinical Safety Considerations.AAPS J. 2015 Sep;17(5):1055-64. doi: 10.1208/s12248-015-9790-0. Epub 2015 May 30.
[3] Endo Y, Mohan N, Dokmanovic M, et al. Mechanisms contributing to ado-trastuzumab emtansine-induced toxicities: a gateway to better understanding of ADC-associated toxicities.Antib Ther. 2021 Mar 16;4(1):55-59.
[4] Mahalingaiah PK, Ciurlionis R, Durbin KR, et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther. 2019 Aug;200:110-125.
[5] Uppal, H, Doudement, E, Mahapatra, K et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine(T-DM1).Clin Cancer Res 2015; 21: 123–33.
[6] Thon JN, Devine MT, Jurak Begon ja, et al. High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)–mediated inhibition of platelet production. Blood 2012; 120: 1975-84.
[7] Zhao H, Gulesserian S, Ganesan SK,et al, Inhibition of megakaryocyte differentiation by antibody-drug conjugates (ADCs) is mediated by macropinocytosis: Implications for ADC-induced thrombocytopenia. Molecular Cancer Therapeutics 16, 1877-1886.[J].
[8] Yan, H, Endo, Y, Shen, Y et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016; 13: 480-90.
[9] Endo, Y, Takeda, K, Mohan, N et al. Payload of T-DM1 binds to cell surface cytoskeleton-associated protein 5 to mediate cytotoxicity of hepatocytes. Oncotarget 2018; 9: 37200-15.
