
▲May 2022, Improved New Drug Trends and Technology Conference·Limited TimeFree Registration Ongoing
Note:This article does not constitute any investment opinions or advice, subject to official/company announcements;This article is only for the introduction of medical health-related drugs, not a recommendation for treatment plans (if involved), and does not represent the platform’s position.Any article reproduction requires authorization.
Introduction
ADC has always been a hot area in drug development due to its precise tumor-targeting killing effect. However, the complex structure of ADC drugs brings certain difficulties to drug design and development. The drugs that have been marketed all have varying degrees of low internalization speed and off-target toxicity, affecting their safety and efficacy, which are all related to the design of ADC drug linkers.
Structure and Mechanism of ADCs
The structure of ADC drugs consists of three parts: the antibody part (Antibody) that specifically recognizes tumor targets, the cytotoxic drug (effective payload/Cytotoxic drug), and the linker (Linker) that connects the effective payload to the antibody.
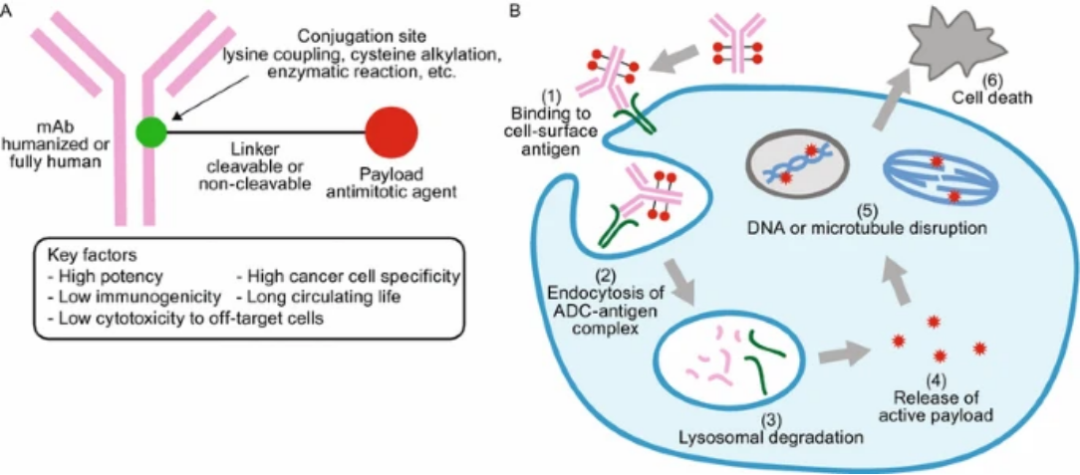
The general mechanism of action of ADCs is shown in Figure B. After the ADC molecule enters the bloodstream, the antibody component of the ADC recognizes and binds to the highly expressed cell surface antigens in target cancer cells. After internalizing the ADC-antigen complex through endocytosis, the complex is processed in the lysosome, where it releases the cytotoxic effective payload (usually anti-mitotic agents) in a bioactive form inside the cell.The released effective payload damages the DNA strand or microtubules, or exerts inhibition on topoisomerase or RNA polymerase, leading to cell death.
ADC Linkers
The design of linkers is the most crucial part of ADC drugs. All aspects of ADC pharmacology may be affected by the specific design of the linker, such as the stability of the drug in circulation, the permeability of tumor cells, the drug-to-antibody ratio (DAR)—the number of effective payload molecules carried by each antibody and their action range, and the bystander effect, etc. Successful construction of an ADC must meet several criteria.
(1) The linker must have sufficient stability in plasma (circulation stability) so that the ADC molecule can circulate in the blood and locate to the tumor site without being cut prematurely. Instability of the linker leads to premature release of toxic effective payloads and unwanted damage to non-target healthy cells, which can lead to systemic toxicity and adverse reactions.
(2) The linker must possess the ability to be quickly cleaved and release free and toxic effective payloads after the ADC internalizes into the target tumor cells.
(3) Another characteristic that needs to be considered in linker design is hydrophobicity. Hydrophobic linkers combined with hydrophobic effective payloads often promote aggregation of ADC molecules.
When designing ADC linkers in practice, several factors need to be considered, including coupling sites, conjugation methods, linker length, linker chemistry, cleavable/non-cleavable linkers, and steric hindrance of proximal linkers.
Structure of Linkers
The way ADC drugs release effective payloads determines their stability in the bloodstream. Based on this classification, linkers can be divided into cleavable linkers and non-cleavable linkers. Cleavable linkers are cleaved in response to extracellular and intracellular environments (pH, redox potential, etc.) or by specific lysosomal enzymes, which allows researchers to estimate the cytotoxic potency of conjugated effective payloads based on known pharmacological parameters.
Cleavable linkers can release while the ADC is not internalized, producing a bystander effect near the tumor target, expanding the killing range. However, cleavable linkers generally have lower stability than non-cleavable linkers and are prone to premature cleavage during circulation, resulting in off-target toxicity; thus, their structures need continuous optimization to achieve a balance between ADC specificity and normal tissue damage. Cleavable linkers can be divided into two main categories: enzymatic and chemical linkers.
Cleavable Linkers—Chemical Linkers
Linkers with Disulfide Bonds
Disulfide bond linkers are glutathione-sensitive conjugates, whose cleavage relies on the high concentration of reducing molecules of glutathione (GSH) in the cytoplasm. GSH is a tripeptide containing thiol, which easily reacts as an S-nucleophile. To ensure the stability of the linker in the bloodstream, a methyl group is often placed next to the disulfide bond, which can resist reductive cleavage in circulation. Upon internalization, the abundant intracellular GSH reductively cleaves the disulfide bond, releasing free effective payload molecules.
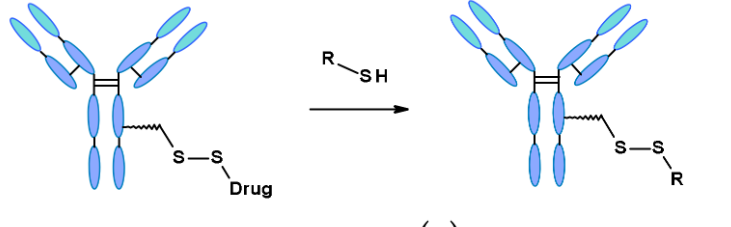
Schematic diagram of disulfide-containing linkers and their reaction with thiols
Disulfide-containing linkers mainly conjugate with maytansine-like effective payloads. The reactivity of disulfide bonds can be adjusted through steric hindrance: α-methyl substitutions significantly affect reduction rates and resistance to thiol-disulfide exchange.
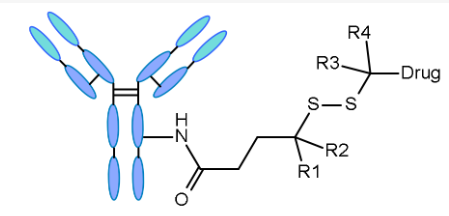
Schematic diagram of egg yolk-like ADC linkers, highlighting the role of α-methyl (R1 to R4)
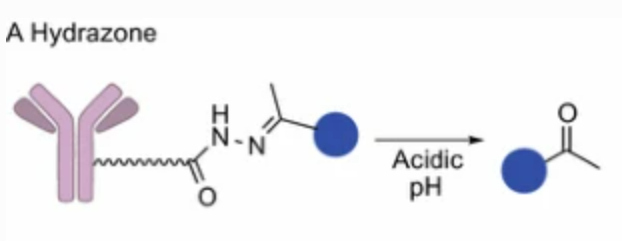
Hydrazone
The first two approved ADCs Mylotarg and Besponsa use hydrazone linkers. Hydrazones are acid-unstable groups, and ADCs with hydrazone linkers transported to acidic endosomes (pH 5.0-6.0) and lysosomes (approximately pH 4.8) will release free drugs through hydrolysis. However, hydrazone linkers have poor stability during circulation (blood circulation), and ADCs using hydrazone as a linker slowly hydrolyze under physiological conditions (pH 7.4, 37°C), leading to slow release of toxic effective payloads.
Non-Cleavable LinkersNon-cleavable linkers in ADCs must be internalized, and the antibody part must be degraded by lysosomal proteases to release active molecules, thus possessing absolute circulation stability. The most representative is SMCC (N-succinimidyl-4-N-maleimidomethyl) cyclohexane-1-carboxylate). The representative drug using SMCC as a linker is TD-M1, whose main tumor metabolite is Lys-SMC-DM1.
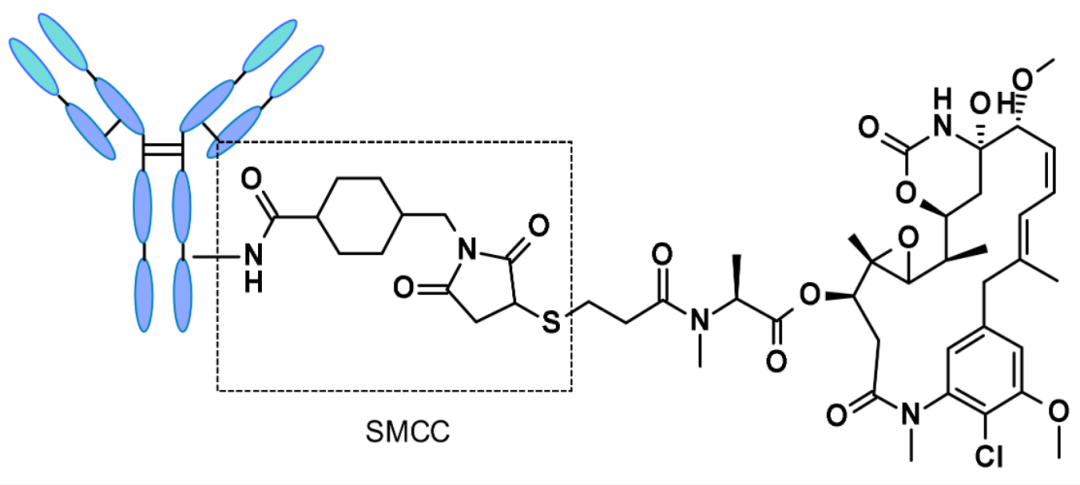
Structure of TD-M1
Cleavable Linkers—Enzymatic Linkers
Substrate Linkers for Cathepsin B
This type of linker is cleaved by the mechanism that, after ADC is internalized through endocytosis and transported to lysosomes, cathepsin B selectively cleaves the linker, releasing the effective payload seamlessly from the ADC, mainly including dipeptide linkers and tetrapeptide linkers.
Dipeptide Linkers originate from the research of cleavable dipeptides as substrates for doxorubicin prodrugs. The P1 position can bind hydrophilic residues, and the P2 position can bind lipophilic residues to increase plasma stability. The PABC part acts as a spacer between the Val-Cit part and the effective payload, limiting the steric hindrance of the effective payload, allowing cathepsin B to exhibit its full proteolytic activity towards linkers attached to bulky effective payload molecules (such as doxorubicin)..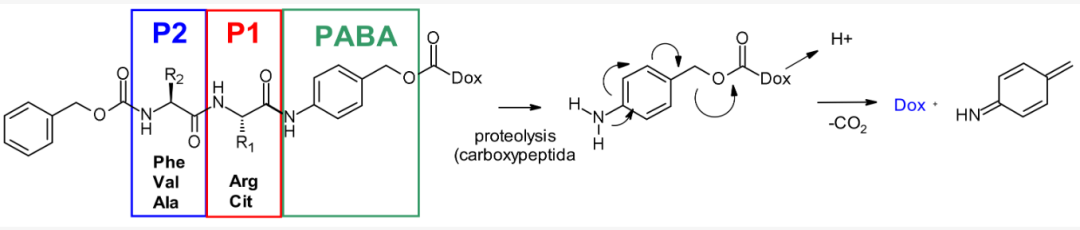
The most commonly used dipeptide linkers include Val-Cit dipeptide (25 clinical) and Val-Ala dipeptide (7 clinical). The buffer stability, cathepsin B release efficiency, cell viability, and histopathological characteristics of the two linkers are comparable. However, due to precipitation and aggregation, Val-Cit is difficult to achieve high DAR, while Val-Ala linkers can achieve a DAR of up to 7.4, but with limited aggregation (<10%). Compared to Val-Cit, Val-Ala has less (non-significant) hydrophobic behavior, making this linker more advantageous in the context of lipophilic effective payloads, such as PBD-dimers. If a hydrophobic spacer (acetyl) is used, then Val-Cit has a higher localized DAR than Val-Ala, and if a small amount of linear PEG12 is added, the difference will not be significant.
Tetrapeptide Gly-Gly-Phe-Gly exhibits all characteristics of a stable and effective cleavable ADC linker and has been successfully applied in Daiichi Sankyo’s Enhertu (DS-8201a), where the linker spacer used is a compact semi-amine.
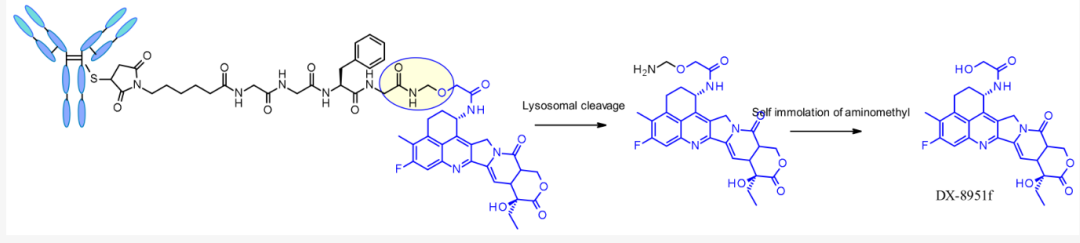 Release mechanism of DS-8201a
Release mechanism of DS-8201a
Pyrophosphate Linkers
Pyrophosphate linkers belong to anionic linkers, possessing higher water solubility and excellent circulation stability compared to traditional linkers. Upon internalization, pyrophosphate undergoes two-step enzymatic linker cleavage through the endosome-lysosome pathway, first releasing effective payload—monophosphate molecules, followed by the release of free effective payloads.
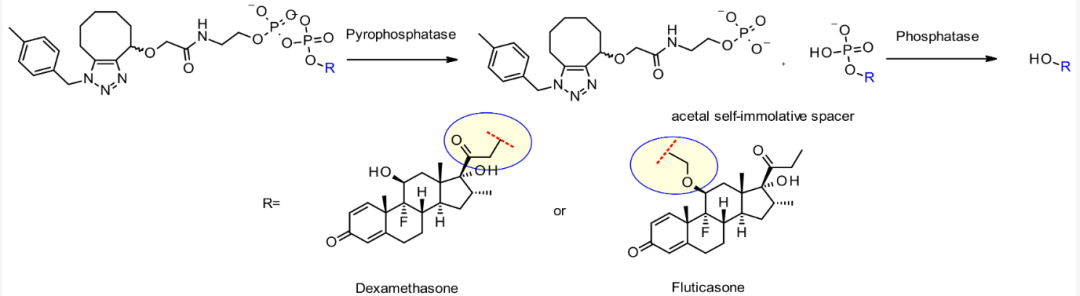
Chemical structure and release mechanism of pyrophosphate linkers
Studies have shown that conjugates constructed with this linker against human CD70 and various glucocorticoids maintain stability in human plasma for 7 days, while being rapidly cleaved to release effective payloads after internalization.
The release rates of effective payloads after internalization of such linkers vary, mainly related to the substituents near the pyrophosphate part, indicating that different structures can be designed to control the release rate.
β-Glucuronidase Substrates
β-Glucuronidase is a type of glycosidase that catalyzes the hydrolysis of β-glucuronic acid residues.This linker has low aggregation levels, high plasma stability, verified cleavage processes in vitro, and strong in vivo efficacy. This linker is also applied to other amine effective payloads, such as CBI microchannel adhesives, camptothecin analogs, and hydroxyl-containing molecules SN38, doxorubicin, and lingzhi protein.
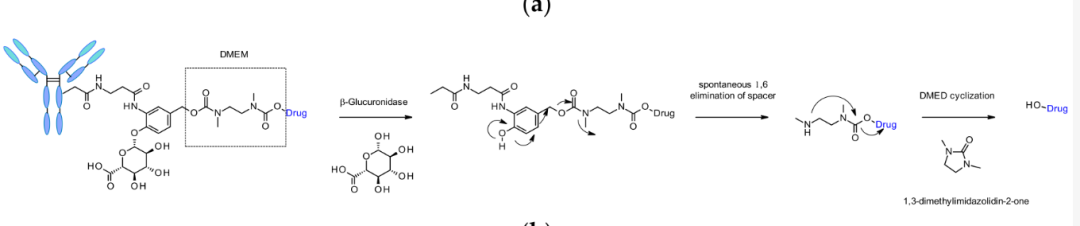
Chemical structure and release mechanism of β-glucuronidase cleavable linkers containing DMED
This linker has good hydrophilicity, making it easier to prepare ADCs with a DAR of 8 compared to linkers sensitive to cathepsin.
β-Galactosidase Substrates
It has been reported that β-galactosidase cleavable linkers are used in ADCs, which incorporate PEG10 spacers. The spacer is substituted with nitro to increase the self-cleavage rate. By analogy with β-glucuronidase linkers, the cleavage mechanism involves hydrolysis of the β-galactosidase part, which imparts hydrophilicity to the chemical precursor. Another advantage is that β-galactosidase is only present in lysosomes, while β-glucuronidase is expressed in lysosomes and also in the microenvironment of solid tumors. Studies have shown that ADCs containing β-galactosidase are more effective in vitro and in vivo than the gold standard Trastuzumab emtansine (T-DM1) in the context of releasing MMAE.
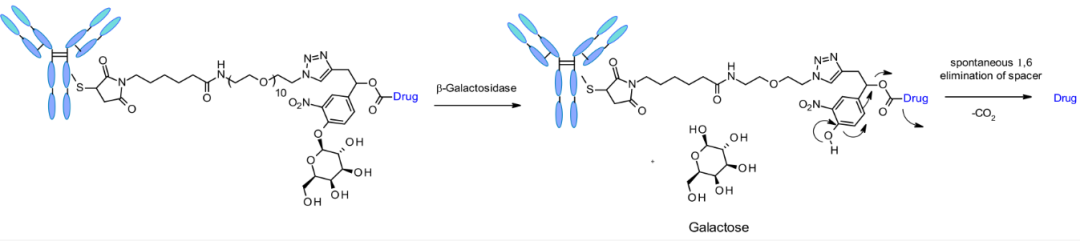
Chemical structure and release mechanism of β-galactosidase linkers.
Sulfatase Substrates
Sulfatase-cleavable linkers serve as glycosidase and (pyro)phosphatase-based linkers, exhibiting good hydrophilicity due to the nature of the target substrate (permanently charged sulfate), while being primarily expressed in lysosomes. Compared to classic cleavable Val-Cit and Val-Ala linkers, sulfatase linkers show similar cellular potency against Her2+ cell lines.
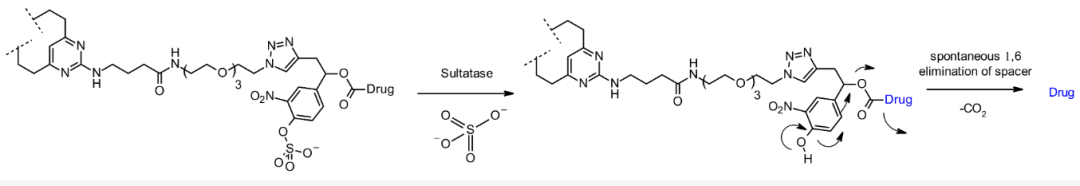
Chemical structure and corresponding release mechanism of sulfatase-cleavable linkers
Conjugation Methods of Linkers
The site of linker conjugation and the conjugation method determine the drug-to-antibody ratio (DAR), and the conjugation site significantly affects the stability and pharmacokinetic-pharmacodynamic characteristics of ADCs—high drug loads often lead to rapid plasma clearance, while ADCs with low DAR exhibit weaker activity.
Chemical conjugation and enzymatic conjugation are currently the two most commonly used methods for linking antibodies and effective payload components.
Chemical Conjugation
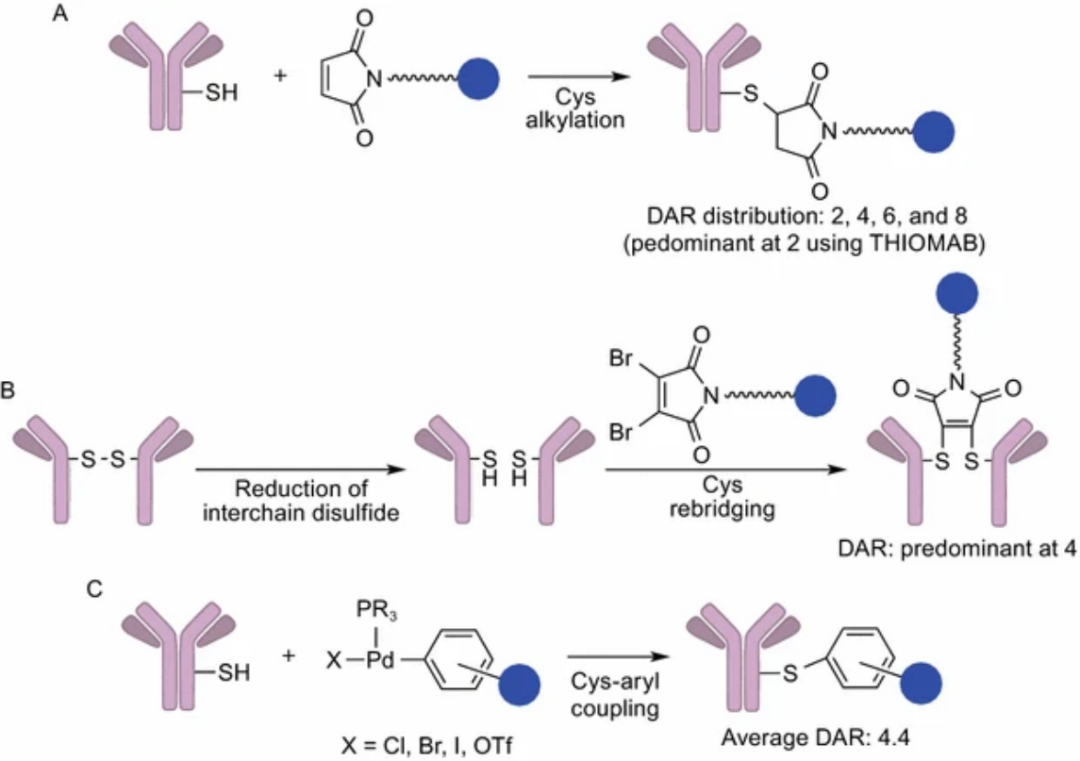
Accessible amino acid residues on the antibody surface are coupled through chemical conjugation between the handle part of the linker and the amino acid residue part of the antibody, eliminating the complexity of determining suitable mutation sites and the potential challenges of expanding and optimizing cell cultures.Early chemical conjugation methods had great randomness in DAR and conjugation sites, resulting in significant differences in the produced ADCs, with insufficient uniform quality.
Lysine Amide Conjugation
Amide conjugation is a major ADC conjugation method, using linkers containing activated carboxylates to connect effective payloads to solvent-accessible lysine residues on the antibody.
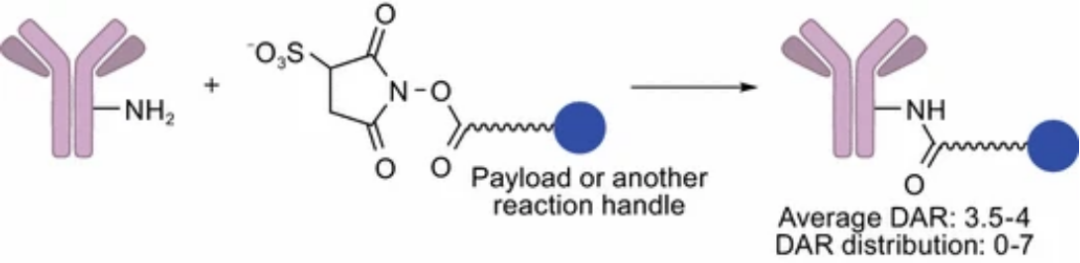
The activated carboxyl part reacts with the lysine residue, leading to the formation of amide bonds between the mAb and the effective payload. Optimized conjugation conditions yield an average drug-to-antibody ratio (DAR) of 3.5–4, with a distribution between 0–7.
Cysteine Conjugation
The specific reaction between cysteine residues on the antibody and thiol-reactive functional groups installed on effective payloads occurs. Antibodies do not have free thiols, and all cysteine residues form disulfide bonds.The advantage is limited conjugation sites, and thiol groups have significant reactivity, thus outperforming lysine conjugation in DAR controllability and heterogeneity.
Enzymatic Conjugation
By using genetically encoded amino acid tags inserted into the antibody sequence, effective payload attachment can be achieved in a very selective manner. These tags are specifically chosen to be recognized by enzymes capable of performing site-specific coupling, such as formylglycine-generating enzyme (FGE), microbial transglutaminase (MTG), sortase, or tyrosinase.
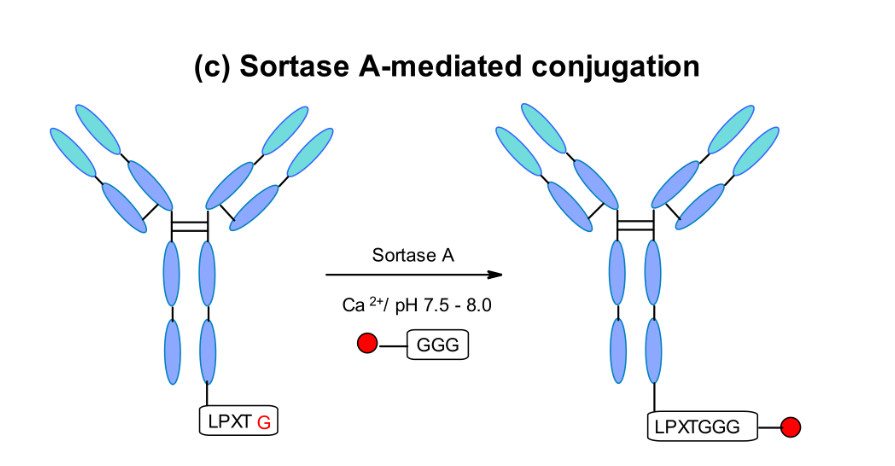
Sortase-Mediated Transduction
NBE Therapeutics has developed a further enzyme-based coupling method using Staphylococcus aureus sortase A-mediated transpeptidation. Their strategy utilizes the enzyme sortase A (Srt A), which cleaves the amide bond between threonine and glycine residues in the LPXTG (X = any amino acid) pentapeptide motif. It then catalyzes the coupling of glycine-derived effective payloads to the newly generated C-terminus, forming peptide bonds at physiological temperatures and pH.This method has been applied to various antibodies, such as anti-CD30 and anti-Her2, where the linkers containing glycine have effective payloads of maytansine and MMAE.
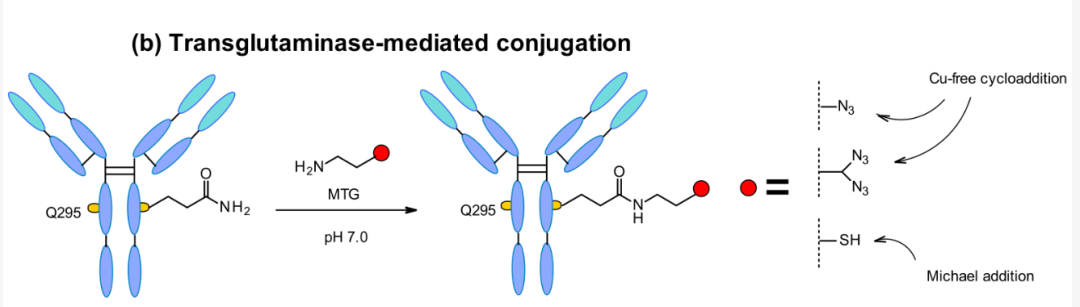
Microbial Transglutaminase-Mediated Transduction
The microbial transglutaminase (MTGase) strategy is also frequently utilized to specifically bind different effective payloads. MTGase catalyzes the formation of peptide bonds between the glutamine side chain at position 295 of deglycosylated antibodies and primary amines of substrates. Compared to other enzymatic strategies, MTG is a flexible technology that does not require peptide donors for conjugation. The acyl receptor only needs to contain primary amines without structural restrictions.
Binding with Glycans
Since human IgG is a glycoprotein, it contains N-glycans located at position N297 in the CH2 domain of each heavy chain. This glycosylation can be utilized as an attachment point for effective payloads of linkers. The distant positioning of glycans from the Fab region reduces the risk of damage to the antibody’s antigen-binding ability after conjugation. Moreover, their different chemical compositions compared to the peptide chains of antibodies allow for site-specific modifications, making them suitable conjugation sites.
Glycan bio-conjugation can be distinguished based on techniques used to target carbohydrates: glycan metabolic engineering, glycosyltransferase treatment, followed by glycan oxidation, endoglycosidase and transferase treatment, and ketone or azide labeling.
OutlookAlthough most ADCs currently in clinical use are based on modifications to natural amino acid sites, the difficulty in obtaining stable DAR ratios at natural amino acid sites has led many companies to focus on the engineering modification of amino acid residues, aiming to achieve more uniformly stable ADCs. For example, the sulfo-Mab technology introduces non-canonical amino acids (ncAA), which provides another possibility for site-specific conjugation. The ncAA technology allows the incorporation of amino acids with unique chemical structures, enabling the selective chemical introduction of linker-effective payload conjugates.References:
[1] The Chemistry Behind ADCs
[2] Antibody-drug conjugates: recent advances in conjugation and linker chemistries[3] The Analysis of Key Factors Related to ADCs Structural Design
Copyright Notice: This article is reproduced from Pharma Financing. If any media or individual does not wish to be reproduced, please contact us, and we will delete it immediately.
<END>
Recent popular activities in the pharmaceutical financing circle, click to view
Offline Activities ↓↓
-
5.31-6.01丨Nanjing·Second Improved New Drug Trends and Technology Conference
-
6.30-7.01 | Guangzhou·Antibody Drug Developers Conference
-
9.07-9.08 | Suzhou Fourth China International Biopharmaceutical & Chemical Pharmaceutical Conference
Online Live ↓↓
-
5.19丨Understanding the Nature of Diseases and Returning to Development Challenges: Is Preclinical Research on PROTAC Difficult?

 Share
Share Like
Like View
View