On November 10, 2025, the research team led by Guan Dongliang from the Shandong Provincial Laboratory of New Drug Creation in Yantai, in collaboration with the research group of Zhang Jinyong from the National Engineering Research Center for Immunological Products at the Army Medical University, published a study titled “Unanticipated Quinoline Modification on Vancomycin as an Effective Strategy to Alter the Antibacterial Profile and Combat Multidrug Resistance” in the Journal of the American Chemical Society.The collaborative team unexpectedly discovered that the quinovancins obtained by simple modification of vancomycin with quinoline groups can effectively overcome bacterial acquired resistance to glycopeptide antibiotics as well as inherent resistance, broadening the antibacterial spectrum of vancomycin and making it effective against multidrug-resistant Enterobacteriaceae.The candidate molecule quinovancin-3d exhibits good drug-like properties and a novel mechanism for penetrating the outer membrane of Gram-negative bacteria, making it a promising representative of a new generation of glycopeptide antibiotics and providing an important emergency drug option for the prevention and treatment of superbug infections.
Antimicrobial resistance (AMR) has become a significant challenge threatening public health and safety worldwide. Statistics from 2019 and 2021 indicate that approximately 5 million deaths globally are associated with bacterial AMR, with Escherichia coli and Staphylococcus aureus being the two pathogens responsible for the highest number of deaths.
As a representative member of glycopeptide antibiotics, vancomycin has been clinically used for nearly 70 years and is regarded as the “last line of defense” against stubborn resistant Gram-positive bacterial infections. Vancomycin exerts its antibacterial effect primarily by binding to the peptidoglycan precursor Lipid II, inhibiting cell wall biosynthesis. However, in recent years, superbugs resistant to vancomycin, such as vancomycin-resistant Staphylococcus aureus (VISA) and vancomycin-resistant Enterococcus (VRE), have emerged, rendering vancomycin ineffective. On the other hand, Gram-negative bacteria are inherently resistant to glycopeptide antibiotics like vancomycin due to the presence of an outer membrane barrier (lipopolysaccharide layer) that prevents vancomycin from reaching the Lipid II target site in the periplasmic space. Therefore, developing new effective strategies to address both acquired and inherent resistance to vancomycin is of significant research importance. While previous studies have reported effective strategies to overcome acquired resistance in Gram-positive bacteria, there are fewer reports on strategies to overcome inherent resistance in Gram-negative bacteria.
Quinoline, as a privileged scaffold, has been widely used in the design of small molecule drugs, imparting various pharmacological activities to derivatives, such as antimalarial, antiviral, antitubercular, antitumor, and anti-obesity effects. Notably, while the application of quinoline in small molecules has been extensively studied, there have been no reports on its introduction into macromolecular peptides or proteins (molecular weight > 1000 Da). In this study, the research team innovatively introduced the 4-amino-7-chloroquinoline privileged fragment from chloroquine into the core structure of vancomycin to design and synthesize a series of vancomycin-quinoline derivatives, collectively named quinovancins. The entire synthesis process is straightforward, efficient, and easily scalable, and the resulting quinovancins exhibit good solubility.
Figure 1 Synthesis route of the final product
Through in vitro antibacterial activity (MIC) testing against various resistant Gram-positive bacteria, the research team found that most vancomycin-quinoline derivatives exhibited antibacterial activity far superior to that of vancomycin, particularly showing 4-256 times increased activity against vancomycin-resistant strains. Furthermore, the research team selected the best compound 3d and tested it against seven clinically isolated resistant Gram-positive bacteria, finding that this molecule demonstrated significantly superior antibacterial activity compared to vancomycin across all types of Gram-positive bacteria. Next, the research team tested four types of Gram-negative bacteria from the ESKAPE group and were pleasantly surprised to find that this class of vancomycin-quinoline derivatives was effective against Escherichia coli, with activity as low as 8μg/mL (~4μM), while the MIC of vancomycin against the tested strains was >128μg/mL, preliminarily indicating the effectiveness of the quinoline modification strategy in overcoming the inherent resistance of Gram-negative bacteria and broadening the antibacterial spectrum. Interestingly, replacing the nitrogen atom in the quinoline with a carbon atom resulted in the loss of antibacterial activity against Escherichia coli, further revealing the important role of quinoline modification. Subsequently, the research team tested compound 3d against more clinically isolated multidrug-resistant Escherichia coli and found that it could broadly overcome the multidrug resistance of Escherichia coli, with MIC as low as 4μg/mL. Considering that Escherichia coli belongs to the Enterobacteriaceae family, the research team also tested another pathogenic bacterium in the Enterobacteriaceae family—Salmonella, and found that 3d exhibited excellent antibacterial activity against various resistant Salmonella strains, with some strains even outperforming ciprofloxacin and polymyxin. This further demonstrates the important role of the quinoline modification strategy in broadening the antibacterial spectrum of vancomycin derivatives, particularly against Enterobacteriaceae bacteria.
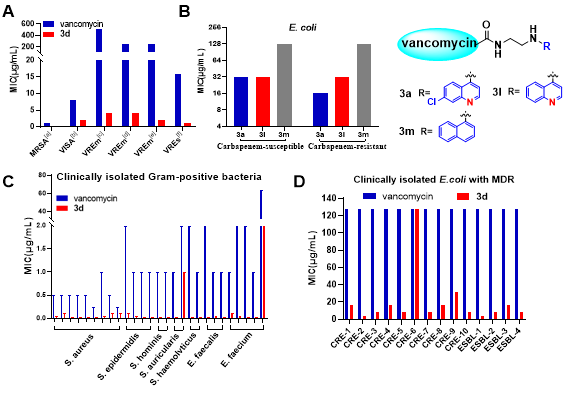
Figure 2 Evaluation of in vitro antibacterial activity
In terms of antibacterial properties, 3d demonstrated inhibitory properties against resistant Gram-positive bacteria and a concentration-dependent rapid bactericidal effect against Escherichia coli. This bactericidal property was further validated by SEM and TEM microscopy images, where swelling of bacterial cells was clearly observed after treatment with 3d, indirectly revealing that 3d inhibited the maturation of peptidoglycan in the periplasmic space.
In the subsequent drug-like property studies, the research team first demonstrated that 3d has excellent in vitro and in vivo safety through cytotoxicity, hemolytic toxicity, and acute toxicity tests. Following this, preliminary pharmacokinetic tests in vivo showed that 3d has superior pharmacokinetic properties compared to vancomycin, and for the first time revealed the important role of the linker in the pharmacokinetic regulation of vancomycin-quinoline derivatives. Then, in the in vivo efficacy evaluation, a single dose (7mg/kg) of 3d achieved 100% survival in mice with MRSA-induced systemic sepsis, while only 20% survival was observed in the vancomycin group. In a mouse model of carbapenem-resistant Escherichia coli (CRE) infection, the 3d treatment group showed significant in vivo efficacy, demonstrating far superior efficacy compared to vancomycin in terms of bacterial colonization in various organs, especially in the kidneys and lungs.
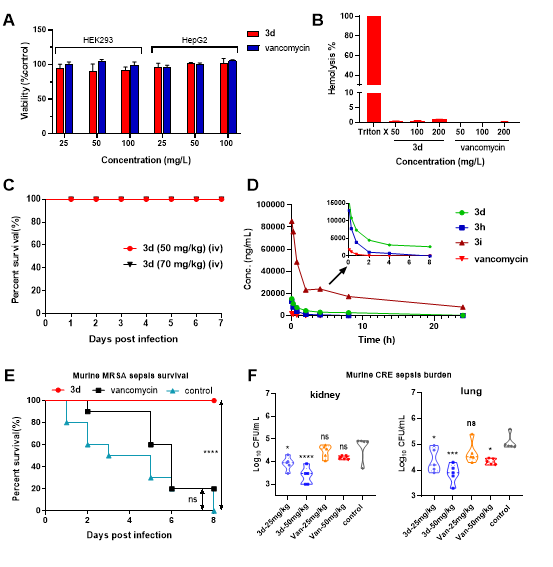
Figure 3 Safety evaluation, in vivo pharmacokinetics, and in vivo pharmacodynamics studies
In the exploration of the mechanism of action, the research team found that 3d significantly enhances the inhibition of cell wall biosynthesis in Staphylococcus aureus and Escherichia coli, leading to a substantial accumulation of park nucleotides, exhibiting a concentration-dependent effect.
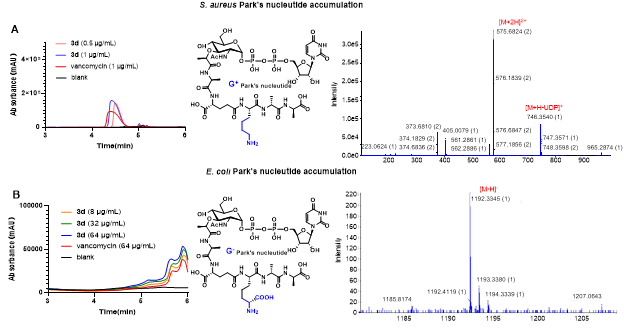
Figure 4 Investigation of cell wall-related mechanisms
In the investigation of the activity on the cell membrane of Gram-positive bacteria, 3d was found to increase the permeability of the cell membrane and induce depolarization, exhibiting strain specificity. Combined with the aforementioned mechanism experiments, it indicates that the role of 3d in overcoming acquired resistance in Gram-positive bacteria arises from both its perturbation of the cell membrane and its enhanced ability to inhibit cell wall biosynthesis. In-depth exploration of the mechanism of action against Escherichia coli revealed that 3d can enter the periplasmic space in a manner that does not disrupt the outer membrane and can further disturb the inner membrane of Gram-negative bacteria, overcoming the inherent resistance of Escherichia coli. This unique and unknown entry mechanism is currently under further investigation.
Additionally, the research team discovered that 3d can increase the accumulation of certain hydrophobic small molecules (e.g., rhodamine B) in the periplasmic space of Escherichia coli, and comparisons with the polymyxin control group in laser confocal images indirectly indicate the non-outer membrane disruptive action of 3d. Interestingly, 3d can enhance the sensitivity of CRE strains to meropenem at sub-inhibitory concentrations, suggesting that the quinoline modification strategy may have sensitizing potential against multidrug-resistant Gram-negative bacteria.
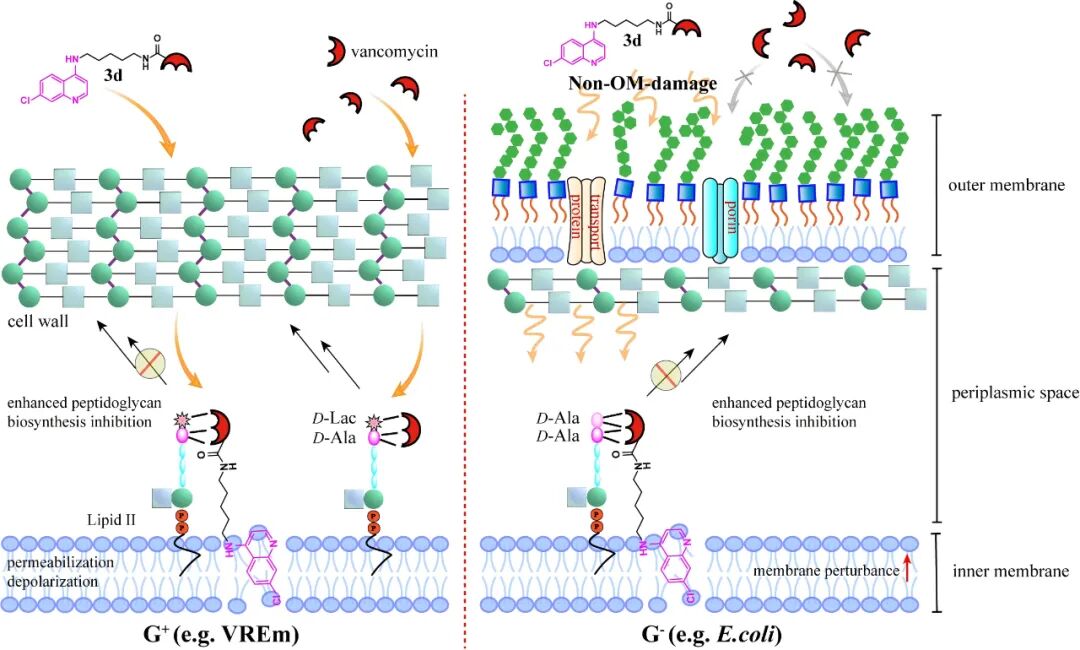
Figure 5 Schematic diagram of the mechanism of action
In conclusion, this study provides a novel strategy based on glycopeptide antibiotics that can help vancomycin overcome acquired resistance in Gram-positive bacteria and, more critically, overcome inherent resistance in Gram-negative bacteria, offering innovative ideas and potential applications for the treatment of super-resistant Gram-negative bacterial infections and opening important directions for addressing the clinical challenges of multidrug-resistant bacteria.
Chang Taopeng, a master’s student from the 2023 joint training program at the Shandong Provincial Laboratory of New Drug Creation in Yantai, is the first author of this paper, while Researcher Guan Dongliang from the Shandong Provincial Laboratory of New Drug Creation and Professor Zhang Jinyong from the Army Medical University are co-corresponding authors. This research work was supported by the National Natural Science Foundation project, the Shandong Provincial Key Research and Development Program (Competitive Innovation Platform) project, the Shandong Provincial Natural Science Foundation project, and the startup fund of the Shandong Provincial Laboratory of New Drug Creation in Yantai. Special thanks to Associate Professor Ren Hao from South China Agricultural University, Associate Professor Zhang Jingwen from Shandong Second Medical University, Professor Zou Quanming from the Army Medical University, Associate Researcher Chen Feifei from the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Professor Feng Meiqing from Fudan University, Professor Feng Xinxin from Hunan University, and Associate Professor Ji Xiaofei from Binzhou Medical University for their guidance and assistance in this research.


 Original link
Original link
https://doi.org/10.1021/jacs.5c14268
Submitted by | Guan Dongliang Research Group
