Boston CollegeProfessor James P. Morken’s team has demonstrated that in the presence of catalytic amounts of Brønsted acid, boron-containing norbornene derivatives undergo regioselective rearrangement to generate boron-containing nortricyclanes. The selective functionalization of boronic esters allows for the acquisition of various multisubstituted tricyclic structures, providing opportunities for broader applications.
Abstract
In the presence of catalytic amounts of Brønsted acid, boron-containing norbornene derivatives undergo a regioselective rearrangement to furnish boron-containing nortricyclanes. The unique stabilization of the putative 2-norbornyl cation by the boron substituent was investigated through both experimental and computational studies. Selective functionalization of the boronic ester product enables access to a diverse array of multisubstituted tricyclic structures, providing opportunities for broader applications.
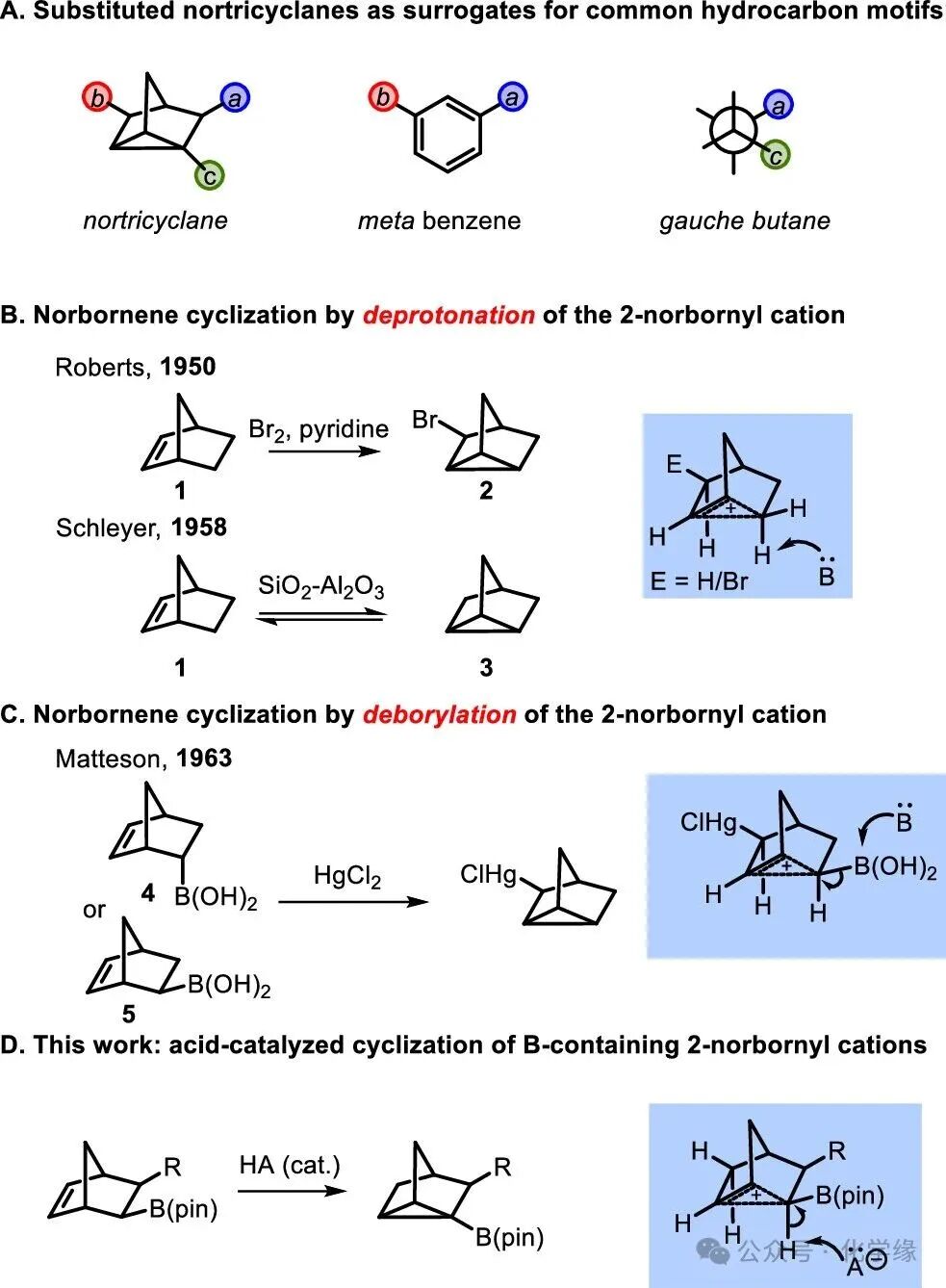
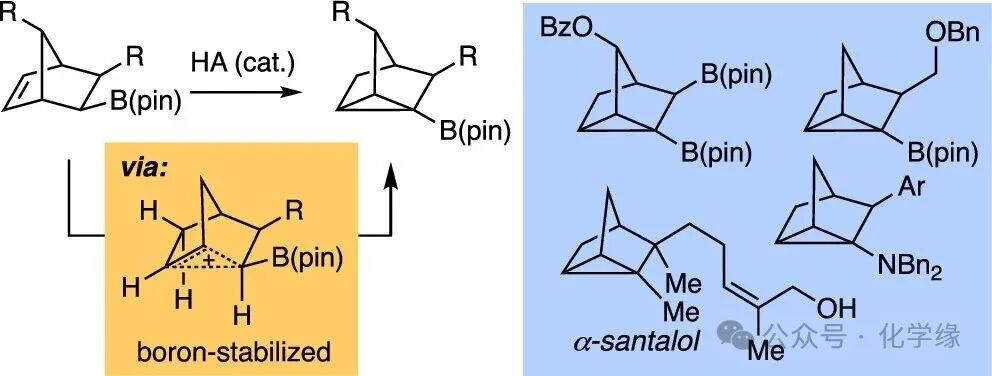
Figure 1 Structures of nortricyclanes and their preparation via 2-norbornyl cation.
Substituted polycyclic scaffolds serve as useful platforms for constructing molecules with spatially defined arrays of substituents. Such architectures find broad applications in drug discovery, receptor design, sensing, and enantioselective catalysis. In this regard, nortricyclanes scaffolds provide numerous opportunities. With substituents connected at positions a and b, the three-dimensional arrangement allows this structure to mimic a para-substituted benzene, while the attachments at a and c exhibit a spatial layout similar to butane. Other substituents attached at different sites provide exit vectors similar to those of other substituted cycloalkanes. A new method for synthesizing boron-containing nortricyclanes is anticipated to enable these compounds to be transformed into various functionalized frameworks with new properties. This article describes an acid-catalyzed rearrangement that appears to proceed through a boron-stabilized nonclassical carbocation intermediate. The reaction occurs under mild conditions, and the presence of boronic esters seems to facilitate a reaction that is incompatible with other substrates, allowing direct access to boron-containing nortricyclanes.
In the 1950s, Roberts demonstrated that bromination in the presence of pyridine could yield 3-bromonortricyclane (2), while Schleyer found that norbornene could be reversibly converted to nortricyclane (3) in the presence of acid catalysts. Both reactions appear to proceed through the generation of a nonclassical 2-norbornyl cation intermediate, which undergoes deprotonation to yield the nortricyclane product. Matteson demonstrated that boron-containing frameworks 4 and 5 could be converted to nortricyclane through a reaction that seemingly involves a substituted 2-norbornyl cation. This article demonstrates that under appropriate conditions, boronic esters can not only accelerate the formation of the 2-norbornyl cation but also promote the deprotonation reaction, yielding novel boron-containing tricyclic products.
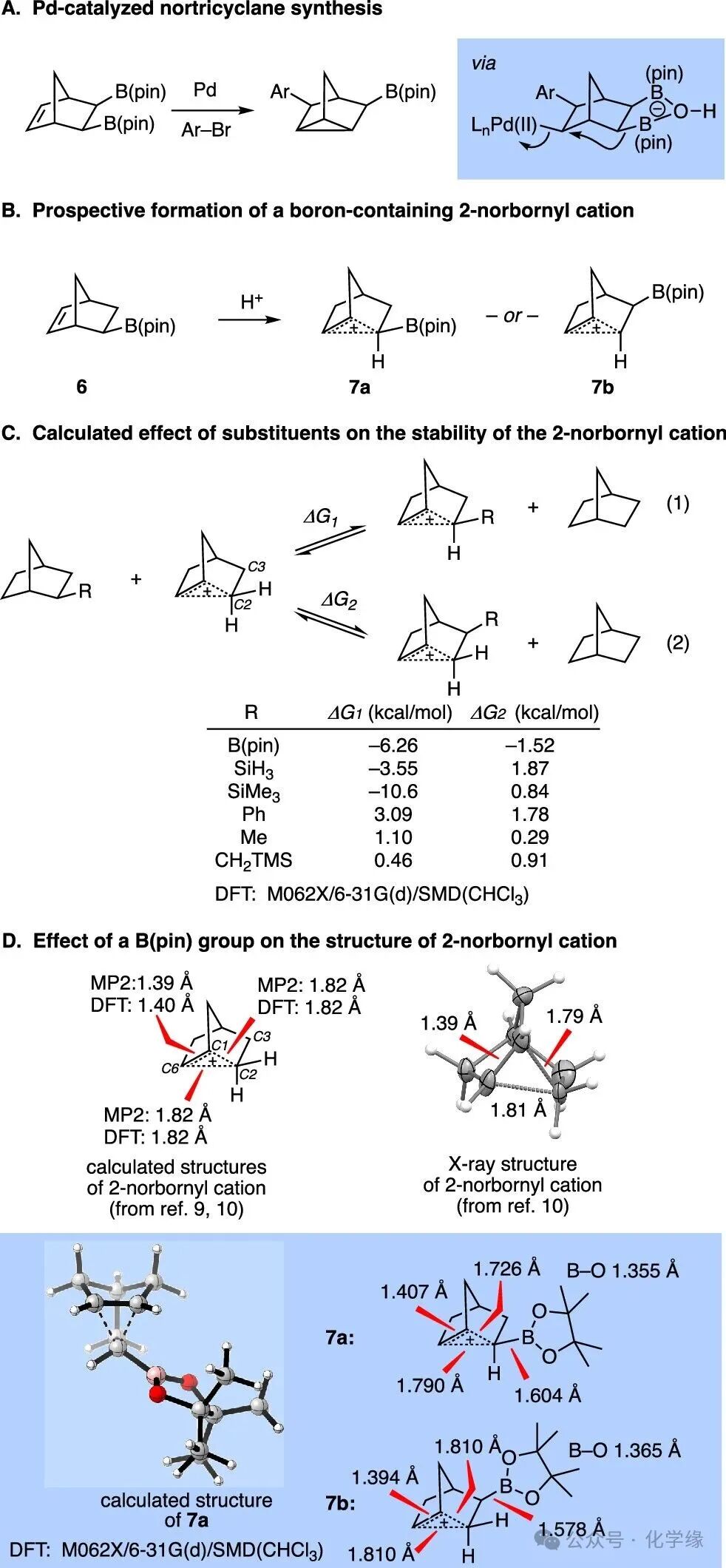
Figure 2 The effect of boronic esters on the stability of the 2-norbornyl cation and DFT calculations.
Related to the above examples, a catalytic enantioselective synthesis of 3,5-disubstituted nortricyclanes (as compounds of para-substituted isomers) was developed through the coupling of boron-containing norbornene and C(sp2) electrophiles. This reaction appears to proceed through carbopalladation, followed by a cross-ring reductive displacement of Pd(II) from the ring system, forming the nortricyclane’s cyclopropane component. Based on this mechanism, and in conjunction with previous studies on the 2-norbornyl cation reactions, it was considered whether boron-containing nortricyclane compounds (such as 6 with Brønsted acid) could be synthesized. Such reactions are expected to generate boron-containing nonclassical carbocations 7a and 7b. Isodesmic-type DFT calculations were performed to analyze these suspected boron-containing carbocations to determine which isomer, 7a or 7b, is more likely to be favored, and to understand whether the presence of boronic esters stabilizes or destabilizes the intermediate cation. Consequently, the free energies of equilibria for equations 1 and 2 were calculated, revealing that the B(pin) group simultaneously attached to C2 and C3 stabilizes the cation, with the boronation at the C2 site having a greater stabilizing effect. Among a series of other substituents, the degree of stabilization appears to correlate with the electron-withdrawing properties of substituents at C2, which are more electron-withdrawing than hydrogen (Me, Ph, CH2TMS) leading to instability relative to the parent 2-norbornyl cation, while electron-donating substituents (SiH3, SiMe3) produced the opposite effect. Compared to the C-B bond distances in 2-borodecane (1.569 Å) and cation 7b (1.578 Å), the C-B bond in 7a is slightly elongated (1.604 Å). These data collectively reflect the enhanced bridging C-C bond in the cation and the weakened C-B σ bond in compound 7a. These findings are consistent with Grob’s experimental observations regarding the substituent effects in the hydrolysis of norbornyl triflates.
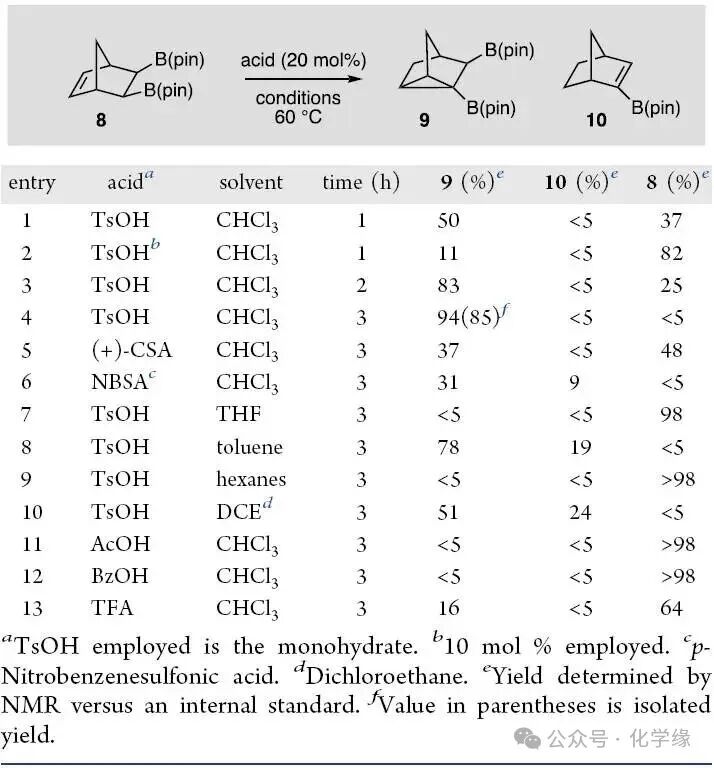
Figure 3 The influence of reaction conditions on the acid-catalyzed cyclization of 8.
Investigating the acid-catalyzed cyclization of boron-containing compound, cesium boronate 8 was treated with para-toluenesulfonic acid monohydrate in chloroform solvent. After 1 hour, no complete conversion was observed, but after stirring for 3 hours, the substrate was consumed, yielding nortricyclane 9 with an 85% yield. It was found that the product of the reaction was not a deboronation during the cyclization process, but rather a deprotonation. Through analysis of the reaction conditions, it was found that treating for 3 hours at 60°C with 20 mol % acid in dichloroethane or chloroform yielded nortricyclane 9, and the only product isomer was obtained.
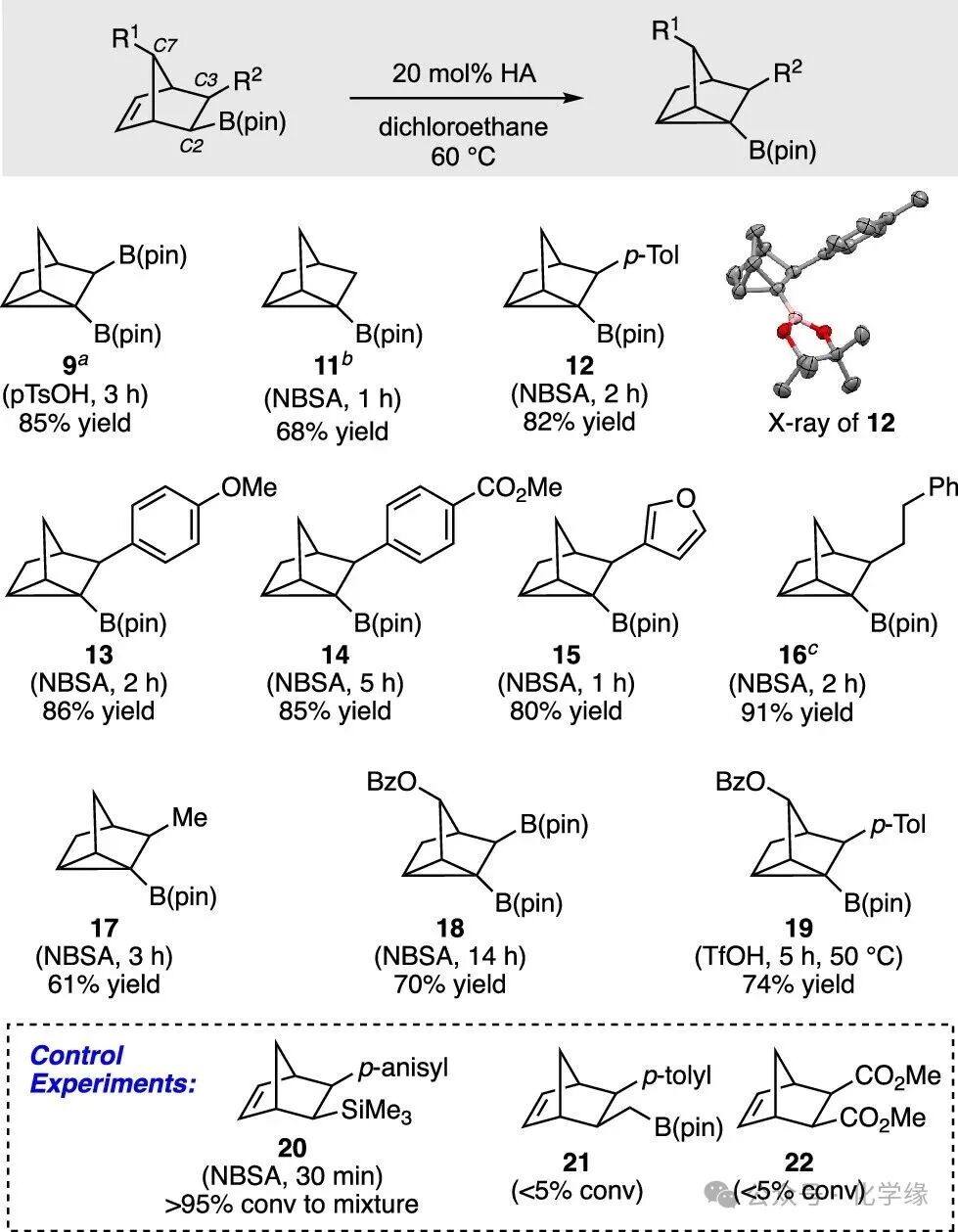
Figure 4 Acid-catalyzed cyclization of boron-containing norbornene derivatives.
Observations of other substrates in the acid-catalyzed rearrangement indicate that by adjusting the reaction conditions, this process can be extended to other boron-containing norbornene scaffolds. The reaction is also effective for monoboronate reactants, yielding 11 with a 68% yield, although this product appears to be less stable than compound 9. With an aromatic substituent at C3, the reaction proceeds efficiently, providing the cyclized products 12–15 as single regioisomers. The regioselectivity of these reactions reflects the superior ability of boron to stabilize the aforementioned intermediate carbocations. Substrates containing C3 alkyl substituents are also reactive substrates and can be cleanly converted to boron-containing nortricyclane (16, 17). Finally, oxygenation at the bridge carbon (C7) was also incorporated into the transformation, yielding more functionalized building blocks (18, 19). Consistent with the calculations that boronic esters may facilitate the formation of intermediate cations, substrates lacking this functional group were unreactive under these conditions (21, 22), while silicon-containing substrates reacted rapidly, although the products were unstable under the reaction conditions.
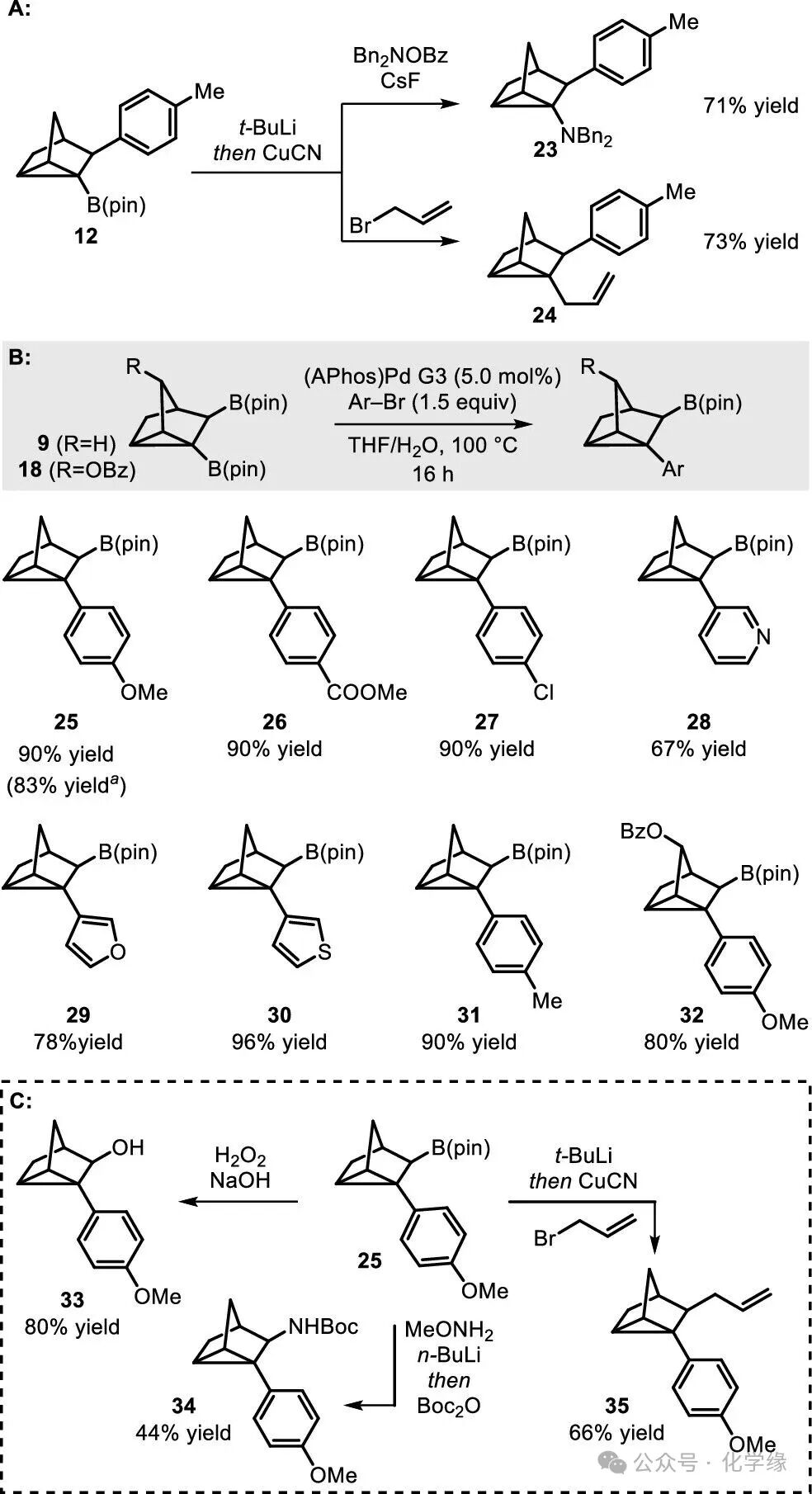
Figure 5 Transformations of boron-containing nortricyclanes.
It was found that copper-catalyzed coupling of monoboronates 12 allows for the replacement of the boron unit with amine (23) or allyl (24). Given that strained cyclopropyl boronates readily undergo ring-opening with electrophilic metal complexes, the high yields of these reactions are particularly notable. In the presence of (APhos)Pd–G3 catalyst and aromatic bromide electrophiles, excellent cross-coupling products 25–31 were obtained from 9, while oxygen-containing substrates 18 were well converted to product 32. Following aromatization, the remaining secondary boronate can undergo transformation to direct oxidation and amination to yield compounds 33 and 34, first activated with tert-butyllithium, followed by copper-catalyzed reaction with allyl bromide to yield compound 35.
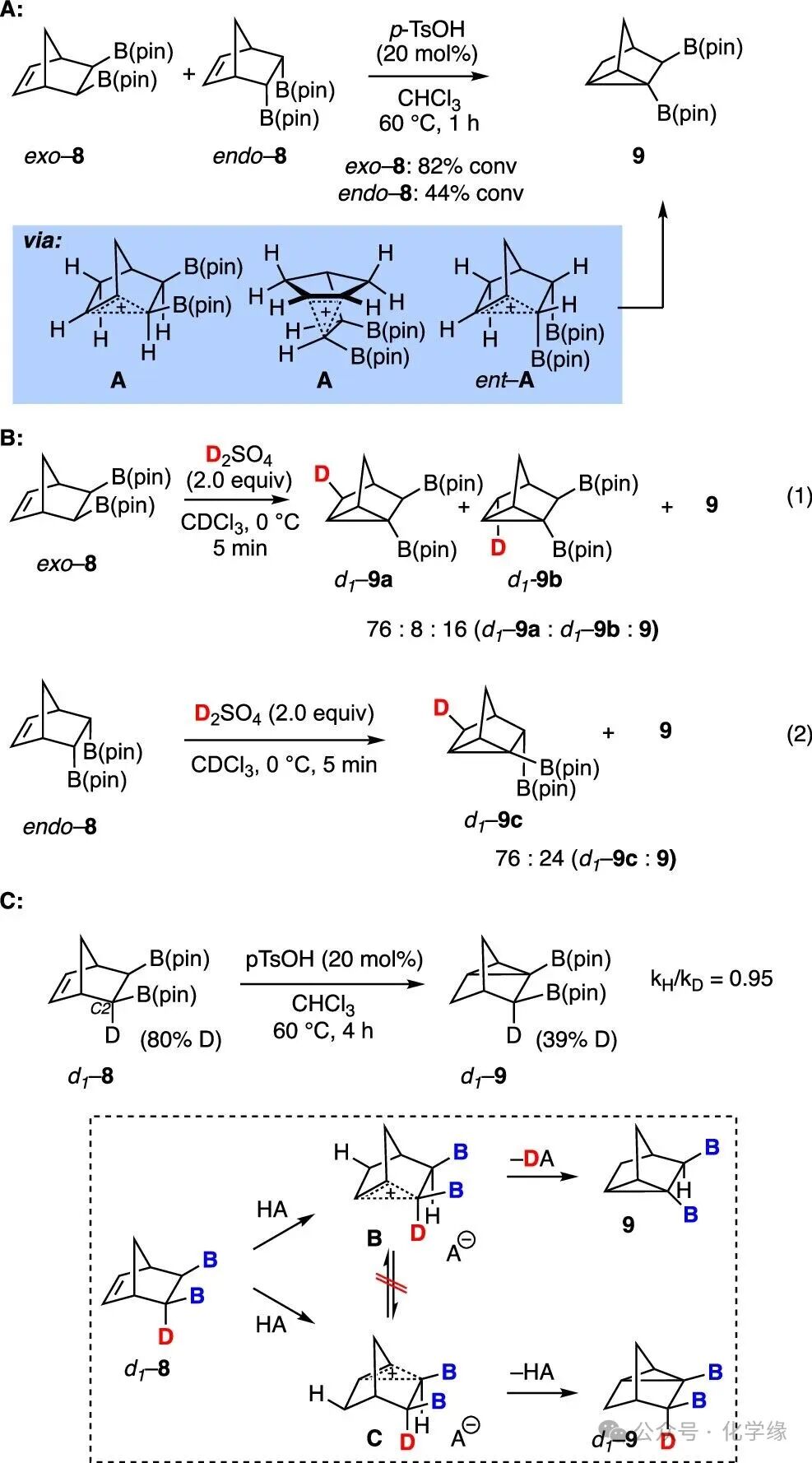
Figure 6 Experimental investigation of the mechanism of acid-catalyzed cyclization of boron-containing norbornene 8.
To explore the mechanism of the acid-catalyzed cyclization reaction, several experiments were conducted. In one set of experiments, the reactivity of 8 was compared between its endo and exo isomers. From the norbornene 8, both exo and endo isomers can generate nortricyclane 9 through the same intermediate carbocation. The difference in reactivity suggests that the rate-limiting step of the rearrangement is the protonation step. Consistent with this observation, treatment of endo and exo 8 with D2SO4 resulted in products containing a single deuterium atom. Although starting from exo-8 was a mixture of stereoisomers. This observation indicates that protonation is irreversible, which is expected when this event is rate-limiting. The final experiment proposed irreversible protonation involving the reaction of isotopically labeled substrate d1–8. The product of this experiment (d1–9) retained 49% of the deuterium label, i.e., kH/kD= 0.95. The lack of a primary kinetic isotope effect also supports the notion that the deprotonation of the intermediate cation occurs faster than known 1,2-hydride transfers—having an activation barrier of 10.8 kcal/mol in the parent norbornyl cation. This would interconvert the intermediate cations B and C.
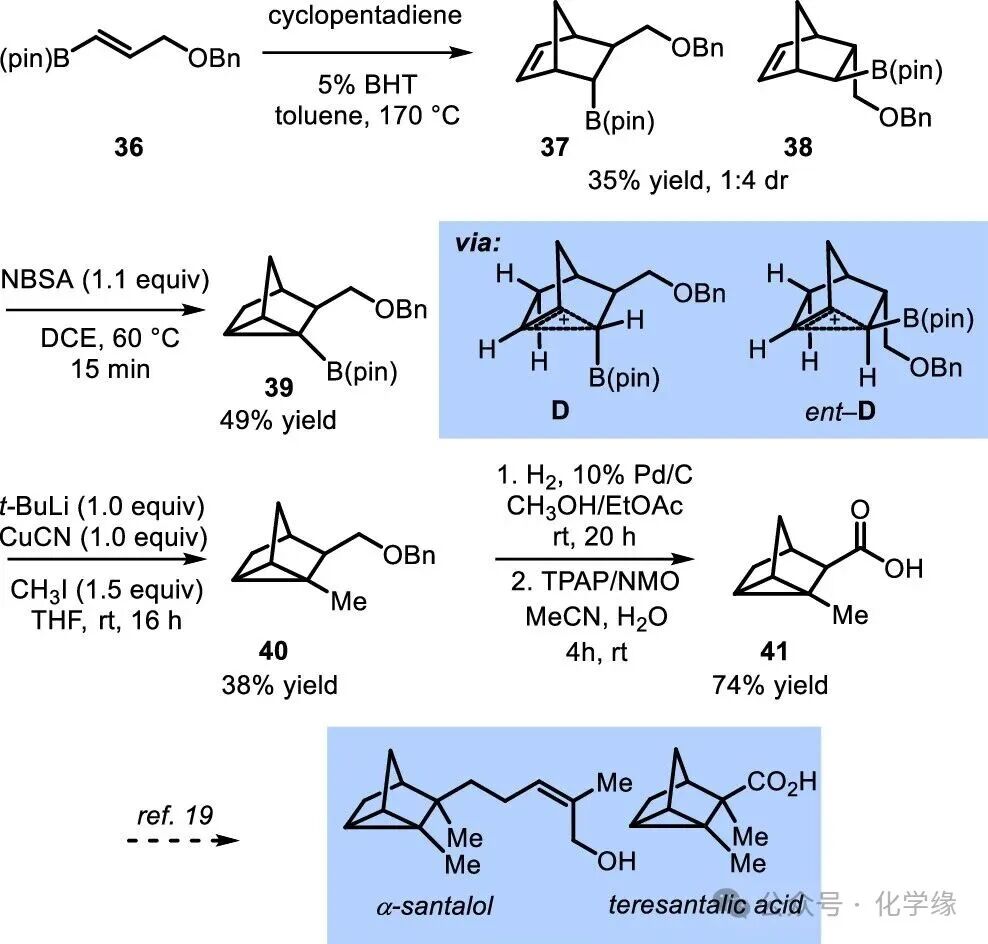
Figure 7 Strategy for synthesizing tricyclic natural products through acid-catalyzed rearrangement of boron-containing norbornene.
The boron-containing nortricyclane products from the aforementioned rearrangement are expected to be used for the construction of ligands and materials, which can also serve as accessible intermediates for constructing substituted tricyclic natural products represented by α-santalol and teresantalic acid. To this end, the reaction of key module 41 was examined. The allyl boronate 36 was transformed into endo and exo adducts 37 and 38 through a Diels–Alder reaction. Although the stereoselectivity of the cycloaddition reaction is not high, both isomers were converted to tricyclic compounds 39 through the intermediate cations D and ent-D. Subsequently, combining with methyl iodide (→40), followed by deprotection and oxidation, yielded the validated intermediates for the synthesis of tricyclic natural products 41.
Conclusion: This study reports the acid-catalyzed rearrangement of boron-containing norbornenes, seemingly operating through boron-stabilized norbornyl cation intermediates. The products of this process are readily accessible and can be modified to provide building blocks with conformational constraints, potentially being utilized in various environments.
Article Information:
Polycyclic Scaffolds through the Intermediacy of Boron-Stabilized Nonclassical Carbocations
Ling Cheng, Mingkai Zhang, James P. Morken*
DOI: 10.1021/jacs.5c15885
We hope this article inspires you, and let us strive together for a better future!
