Hello everyone, today I would like to share an article published in the Journal of the American Chemical Society, titled “Observation of *N2H3 Intermediates by In Situ Electrochemical SERS: New Pathway of Ammonia Oxidation on High-Durability PtIr Catalysts“. The corresponding author of the article is Professor Xie Wei from Nankai University.

Abstract
The electrochemical ammonia oxidation reaction (AOR) is a core technology for sustainable on-site hydrogen production and clean energy conversion. Although platinum-iridium (Pt-Ir) alloy catalysts are promising for addressing the rapid deactivation issue of pure Pt systems, the atomic-level mechanism behind their superior stability remains unclear. This study employs in situ electrochemical surface-enhanced Raman spectroscopy (EC-SERS) to directly track the evolution of intermediates on Pt and PtIr catalysts during the AOR process. The study finds that on pure Pt electrodes, increasing the potential drives *NH2 to sequentially oxidize to *N3–, ultimately forming surface-bound *NOx species, which lead to deactivation of active sites and performance degradation; whereas the PtIr alloy avoids this detrimental pathway through a unique *N2H3 center mechanism—on the Pt-Ir synergistic catalytic sites, *NH couples with *NH2 intermediates to generate *N2H3, which is further dehydrogenated to form N2, without producing toxic byproducts. Therefore, PtIr alloy catalysts exhibit significantly better durability than monometallic Pt catalysts. This study reveals the important value of directly observing key reaction intermediates through in situ spectroscopy for elucidating catalytic mechanisms.
Introduction
Ammonia (NH3) has become a promising hydrogen carrier due to its high hydrogen storage capacity of up to 17.6 wt.%. Its advantages also include ease of liquefaction for safe storage and transportation, and zero carbon emissions after complete dehydrogenation, making it a cornerstone of next-generation clean energy technologies. However, the practical application of ammonia in the hydrogen economy faces critical bottlenecks: traditional thermal catalytic decomposition requires high temperatures (> 500 °C), severely limiting its compatibility with mainstream hydrogen applications. In contrast, the electrochemical ammonia oxidation reaction (AOR) can produce hydrogen under mild conditions, with a theoretical potential of 0.059 V vs RHE, which is much lower than that of water electrolysis (1.23 V vs RHE), making it an energy-efficient chemical reaction. However, the multi-step proton-coupled electron transfer (PCET) process leads to kinetic lags, resulting in suboptimal starting potentials in practical systems. Platinum (Pt) based catalysts are recognized as benchmark anode materials for AOR due to their low starting potential and high current density. However, according to the Oswin-Salomon mechanism, irreversible adsorption of *N species can cause chemical deactivation, fundamentally affecting their operational stability. Recent studies indicate that accumulated nitrogen oxides (*NOx) byproducts may become major poisons through strong metal-adsorbate interactions.
Alloying Pt with transition metals can effectively alleviate surface deactivation, with iridium (Ir) standing out due to its compatible valence electron configuration and chemical synergistic effects with Pt. Experimental results confirm that PtIr alloy catalysts not only lower the starting potential for AOR but also exhibit superior anti-poisoning capability and long-term durability compared to pure Pt. Despite these advancements, little is known about the dynamic structural evolution of Pt-based catalysts during the AOR process. Due to the transient nature of reaction intermediates, weak spectral signals, and the complexity of the electrochemical environment, a critical knowledge gap remains regarding the correlation between interfacial species transformation and catalytic activity. Traditional characterization techniques struggle to capture real-time surface evolution, necessitating the development of advanced in situ analytical methods. Surface-enhanced Raman spectroscopy (SERS) combines micron-scale spatial resolution, signal enhancement of 106-108, and molecular fingerprint specificity through localized surface plasmon resonance (LSPR) effects, providing a revolutionary breakthrough for interfacial reaction analysis.
This study employs in situ electrochemical SERS (EC-SERS) technology to reveal the dynamic evolution of interfacial species on the surface of PtIr alloy nanoparticles during the alkaline AOR process (Figure 1). Through comparative experiments and isotopic labeling studies, this research uncovers the differentiated reaction pathways of Pt and PtIr systems: pure Pt surfaces exhibit intermediates *N3– and *NOx that lead to catalyst poisoning, while the PtIr alloy specifically detects a novel *N2H3 adsorbate formed through the N-N coupling of *NH and *NH2. Theoretical analysis indicates that this pathway is regulated by the unique ten-electron interaction rules between the Pt-Ir synergistic catalytic sites and the adsorbates, effectively avoiding the accumulation of toxic intermediates. This breakthrough mechanism not only provides atomic-level insights into AOR catalysis but also lays the theoretical foundation for designing high-performance anti-poisoning electrocatalysts.
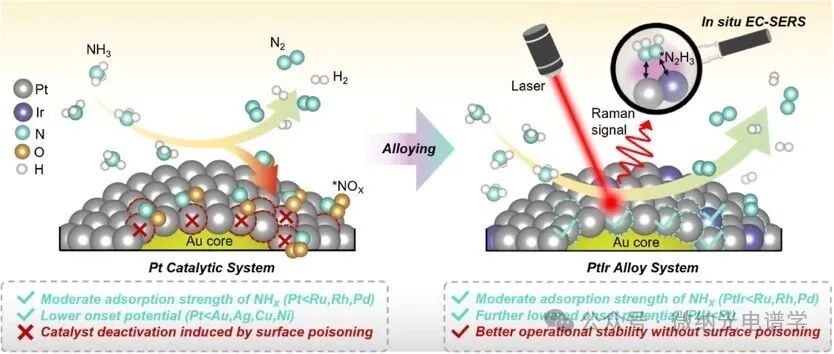
Figure 1. Schematic of in situ EC-SERS during the AOR process on Pt-based catalysts. During the electrocatalytic AOR process, the active sites of the Pt catalyst (red circles) are occupied by *NOx intermediates (*i indicates adsorbed species), making them difficult to desorb. In contrast, the PtIr alloy catalyst exhibits excellent resistance to *NOx formation, ensuring the accessibility of active sites (cyan circles) throughout the catalytic cycle.
Results and Discussion
Characterization of Pt-based Catalysts and Construction of EC-SERS Device
Iridium (Ir) content varying platinum (Pt) nanoparticles (NPs) and platinum-iridium (PtIr) nanoparticles were prepared using a thermal decomposition method. The content of Pt and Ir elements was determined by inductively coupled plasma mass spectrometry, confirming that Ir was successfully incorporated into the alloy. X-ray photoelectron spectroscopy (XPS) analysis showed a systematic high binding energy shift in the Pt 4f spectrum, while the Ir 4f spectrum exhibited a binding energy downshift, indicating the presence of electron transfer from Pt to Ir in the PtIr nanoparticles, consistent with previous reports. As shown in Figure 2a, the X-ray diffraction (XRD) pattern of PtIr nanoparticles shows two characteristic peaks at 39.6° and 46.2°, corresponding to the (111) and (200) crystal planes of face-centered cubic (fcc) structure. The observed peak shifts confirm the formation of the alloy, consistent with the results of JCPDS 88-1728 standard card. The high-resolution transmission electron microscopy (HRTEM) image in Figure 2b shows that Pt0.88Ir0.12 nanoparticles have a clear cubic morphology with an average edge length of 9.70 ± 0.04 nm. Furthermore, the incorporation of Ir into Pt leads to a lattice contraction of the (200) crystal plane, with the interplanar distance decreasing from 0.197 nm to 0.194 nm (Figure 2c).
The electrochemical testing of Pt-based catalysts indicates that the PtIr alloy system exhibits superior performance and anti-poisoning capability in the ammonia oxidation reaction (AOR). Pt0.88Ir0.12 shows the highest peak current density and lowest impedance at 0.7 V vs RHE. Stability tests reveal significant differences in catalyst durability: pure Pt experiences over 90% current decay by the 10th cycle, while the Pt0.88Ir0.12 system demonstrates excellent anti-poisoning performance—after an initial drop of 50% during the activation phase (1-5 cycles), the current density recovers and stabilizes at 17 mA cm-2 (Figure 2d-f). Chronoamperometry (CA) tests further indicate that after accounting for the initial decay of the non-Faradaic double layer current, the current density of the PtIr alloy remains consistently higher than that of pure Pt and pure Ir, confirming its superior durability (Figure 2g).
This study prepared Au@Pt and Au@Pt0.88Ir0.12 core-satellite superstructures for in situ electrochemical surface-enhanced Raman spectroscopy (EC-SERS) monitoring of the AOR process. The near-field electric field distribution of the nanoparticles at an excitation wavelength of 632.8 nm was evaluated through three-dimensional finite-difference time-domain (FDTD) simulations (Figure 2h). Compared to the negligible electric field enhancement of the PtIr nanoparticle array, the electric field strength within the superstructure gaps is significantly enhanced, with a relative |E|2/|E0|2 value exceeding 20. Using a self-made electrochemical cell with a monolayer of nanoparticles modified working electrode, this study conducted in situ EC-SERS tests on the AOR process in 1 M NH3 and 1 M KOH electrolytes (Figure 2i).
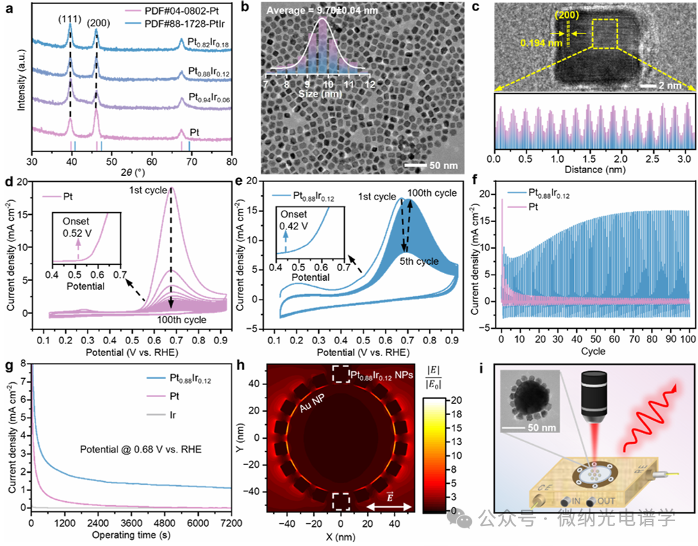
Figure 2. (a) X-ray diffraction (XRD) patterns of Pt and PtIr nanoparticles with varying Ir content. (b) High-resolution transmission electron microscopy (HRTEM) image and large-area distribution of Pt0.88Ir0.12 nanoparticles. (c) The lattice spacing of a single Pt0.88Ir0.12 nanoparticle is 0.194 nm, corresponding to the (200) crystal plane (upper image); the diffraction fringes (lower image) correspond to the area within the yellow dashed box. (d,e) Cyclic voltammetry curves of Pt and Pt0.88Ir0.12 electrocatalysts in 1 M NH3 and 1 M KOH aqueous solutions during the ammonia oxidation reaction (AOR) (100 cycles), with a scan rate of 50 mV/s. The initial cycle starting potentials are 0.52 V (Pt) and 0.42 V (Pt0.88Ir0.12). (f) Extracted cyclic current density curves of Pt and Pt0.88Ir0.12 electrocatalysts during the AOR process. (g) Chronoamperometry (CA) curves of different catalysts at 0.68 V vs RHE, with testing conditions in 1 M NH3 and 1 M KOH solutions. (h) Finite-difference time-domain (FDTD) simulation showing the electric field distribution under parallel illumination of 632.8 nm laser (XY plane) on Au@PtIr core-satellite superstructures (80 nm Au core; 9 nm PtIr satellite particles; 0.8 nm spacing between Au and PtIr). (i) Schematic of the in situ electrochemical surface-enhanced Raman spectroscopy (EC-SERS) testing device, with the upper left inset showing the HRTEM image of Au@Pt0.88Ir0.12 core-satellite superstructure.
EC-SERS Characterization Study of Pt Catalysts
This study collected in situ EC-SERS spectra of the AOR process on Pt NPs with a potential increment of 50 mV (0.22-0.92 V vs RHE) (Figure 3a, b). According to existing studies, the characteristic peaks at 2095 cm-1 and 3251 cm-1 correspond to the stretching vibrations of C-O adsorbed at the top site and N-H stretching vibrations of adsorbed NH3, respectively. The three signals at 324, 1393, and 2017 cm-1 exhibit significant Stark shifts with potential changes, and these signals did not appear in control experiments with pure Au and 1 M KOH, indicating that they correspond to nitrogen-containing intermediate species chemically adsorbed on the surface of Pt NPs. Among them, the peaks at 2017 cm-1 and 1393 cm-1 correspond to the asymmetric N-N-N stretching vibration of azide (*N3–) and *NOx species.
Based on in situ SERS evidence, this study proposes a dual-pathway mechanism for AOR on Pt electrodes (Figure 3c, d). Pathway I (main pathway): As the potential increases, the N-H vibration intensity decreases, accompanied by the formation of NH2, which is subsequently oxidized to *N3– through a six-electron transfer step; this species further reacts at the oxidation peak potential of AOR to generate N2. Pathway II (side pathway): The competitively generated NO is subsequently oxidized to NO2– and NO3– at higher potentials. Notably, these nitrogen oxides have a strong affinity for the Pt surface, which explains the rapid deactivation phenomenon observed in experiments.
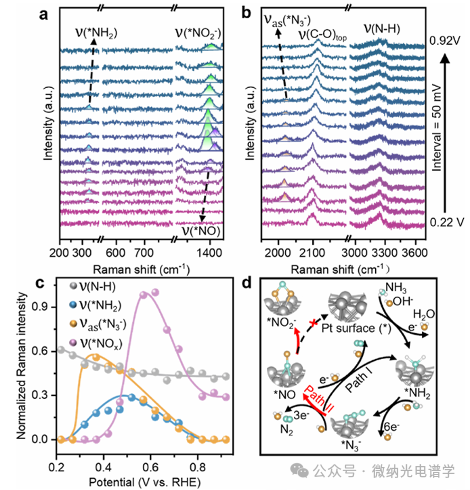
Figure 3. (a,b) In situ EC-SERS spectra of the AOR process on Pt nanocubes (composed of six (200) crystal planes) in 1 M NH3 and 1 M KOH solutions, scanned from 0.22 V to 0.92 V vs RHE with a step size of 50 mV. (c) Normalized SERS intensity changes of four vibrational modes at different potentials. (d) Schematic diagram of the alkaline AOR reaction mechanism on pure Pt surface: *N3– oxidized to generate *NOx species strongly anchors on Pt active sites (Pathway II), inhibiting the N2 generation pathway (Pathway I) through competitive site occupation.
EC-SERS Characterization Study of Pt0.88Ir0.12 Catalysts
The in situ EC-SERS analysis of AOR on Pt0.88Ir0.12 catalysts reveals key mechanistic differences that enhance durability (Figure 4a-c). At the initial potential, Raman modes corresponding to C-O and N-H bonds can be observed. Notably, the SERS signal matching the NH2 vibration at 324 cm-1 persists in the low potential range in the alloy system, which is not observed in pure Pt NPs. The coexistence of *NH2 and *NH species at the initial potential correlates with the lowering of the starting potential in cyclic voltammetry tests of Pt0.88Ir0.12, indicating that the alloy catalytic sites facilitate the kinetics of proton-coupled electron transfer (PCET). Interestingly, as the potential increases, the initial SERS signals gradually diminish, while two new vibrational modes appear at 728 cm-1 (0.32 V) and 606 cm-1 (0.57 V) (Figure 4b, d).
Due to the larger atomic mass of metal atoms, it is challenging to distinguish the mass effect calculations of Pt-N and Ir-N vibrational modes, making it difficult to determine the specific metal adsorption sites of intermediates at 324, 606, 686, and 728 cm-1. However, the *N2Hn intermediates are only present on the surface of Pt0.88Ir0.12 and not observed in pure Pt or Ir systems, indicating that the unique reaction pathway of the alloy may be a key factor in enhancing catalytic stability (Figure 4e).
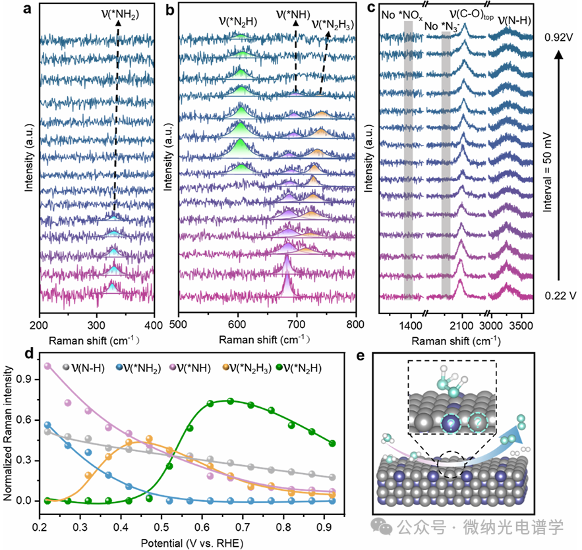
Figure 4. (a-c) In situ EC-SERS spectra of the AOR process on Pt0.88Ir0.12 nanocubes (composed of six (200) crystal planes) in 1 M NH3 and 1 M KOH solutions, with a potential scan range of 0.22 to 0.92 V vs RHE and a scan step of 50 mV. (d) Normalized SERS intensity changes of five vibrational modes at different potentials. (e) Schematic diagram of the AOR reaction mechanism on Pt0.88Ir0.12 (200) catalyst surface, where the specific adsorption sites of *N2H3 species remain unclear.
In-depth Analysis of the AOR Mechanism on Pt0.88Ir0.12 under Alkaline Conditions
This study further elucidates the structure-activity relationship of AOR on Pt0.88Ir0.12 NPs through DFT calculations. Thermodynamic stability studies indicate that the system exhibits an adsorption evolution mechanism mediated by *N2H3 intermediates. As shown in Figure 5a, the potential-determining step involves the N-N coupling of bridge-adsorbed *NH2 and *NH occurring on adjacent Pt-Ir atoms to form *N2H3. The formation energy barrier of *N2H3 at the Pt-Ir synergistic catalytic sites (0.90 eV) is lower than that at adjacent Pt-Pt sites (1.14 eV) by 0.24 eV, revealing the dual-metal synergistic regulation of intermediate adsorption in the PtIr alloy system. This study extends the ten-electron counting rule proposed by Reocreux et al. to analyze the dynamic intermediate transformation process. When this condition is met, the catalytic system is in a stable state, where νM represents the valence electron count of the transition metal, and k represents the number of electrons involved in the interaction between the adsorbate and the transition metal d orbitals.
Figure 5b illustrates the bridging adsorption configurations and electronic sharing modes of *NH2 and *NH on Pt0.88Ir0.12 NPs: the three electrons of *NH2 share one with Pt1, one with Pt2; the four electrons of *NH are more inclined to share one with Pt3 and one with Ir, retaining one lone pair electron. The analysis indicates that the maximum νPt 2 + κNa value (12) corresponds to the weakest Pt2-Na interaction, making it easy to break during subsequent N-N coupling; while the value of νIr + κNe = 10 indicates the strongest chemical adsorption of nitrogen-containing species at the Ir site.
Figure 5c reveals the electronic distribution characteristics of *N2H3 at the Pt-Ir synergistic sites: its formation primarily occurs through the breaking of the weakest Pt2-Na bond, and the participation of Ne lone pair electrons in forming N-N double bonds. In contrast, Figure 5d shows that the adsorption of *N2H3 at adjacent Pt sites requires the simultaneous breaking of relatively stable Pt1-Na and the most stable Ir-Ne bonds, which violates the ten-electron counting rule, thus requiring a higher formation energy. Therefore, the electronic coupling of Ir valence orbitals with intermediates fundamentally alters the AOR reaction pathway.
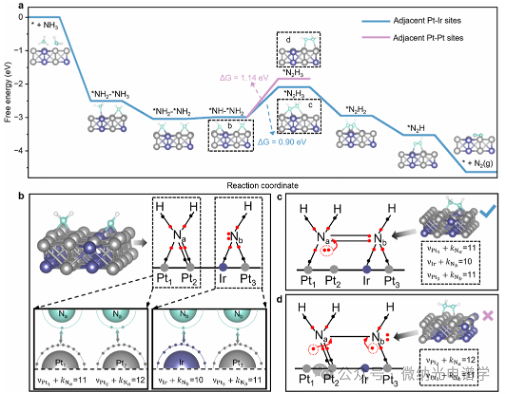
Figure 5. (a) Gibbs free energy diagram of Pt0.88Ir0.12 (200) crystal plane under pH=14 and standard potential (U = 0 V vs RHE) (the structure within the black dashed box will be discussed in detail in (b-d)). (b) Schematic diagram of the electronic interaction of *NH-*NH2 with metal surface electrons: the upper right shows the Lewis structure (red dots indicate electrons involved in bonding with metal intermediates), and the bottom shows the outer orbital arrangement. (c, d) Comparative analysis of the adsorption configurations of *N2H3 at Pt-Ir synergistic sites (c) and Pt sites (d), where the intermediate is formed through the N-N coupling of *NH2 and *NH.
Figure 6 illustrates the AOR reaction mechanism of Pt0.88Ir0.12 electrode in alkaline electrolyte, integrating experimental and theoretical evidence. The reaction pathway consists of seven steps: (1) After the NH3 molecule adsorbs on the surface of Pt0.88Ir0.12 NPs, it undergoes continuous dehydrogenation through two proton-coupled electron transfer (PCET) steps (steps (1-2)), forming complementary *NH2 and *NH species on the PtIr alloy surface; (2) *NH2 and *NH couple at the Pt-Ir synergistic catalytic sites to generate the thermodynamically more favorable *N2H3 intermediate (step (3)), which is significantly different from the pathway generating *N3– in pure Pt systems; (3) Subsequent stepwise dehydrogenation ultimately leads to the formation and desorption of N2 (steps (4-7)), completing the catalytic cycle. This mechanism confirms the hypothesis that increasing OH– concentration can promote dehydrogenation steps, thereby enhancing catalytic activity. This pathway centered on *N2H3 strongly validates the partial dehydrogenation theory proposed by Gerischer and Mauerer. By inhibiting the formation and accumulation of nitrogen oxides on the electrode surface (the main cause of pure Pt catalyst deactivation), the PtIr alloy achieves excellent operational stability.
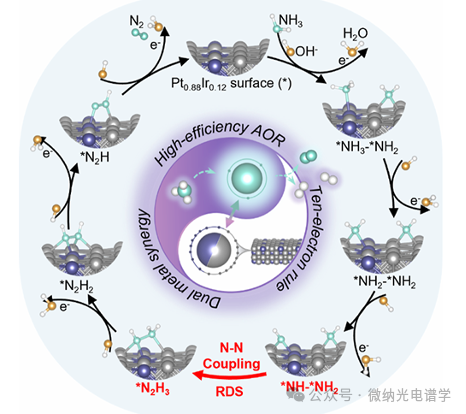
Figure 6. Schematic of the AOR reaction mechanism on Pt0.88Ir0.12 electrode under alkaline conditions (red arrows indicate rate-determining steps). The unique electronic interactions of intermediates with the alloy surface promote the N-N coupling process, effectively suppressing the formation of toxic *NOx species, thereby achieving efficient and stable ammonia oxidation reactions.
Conclusion
In summary, the bifunctional Au@(Pt/PtIr) superstructure system designed in this study successfully elucidates the mechanism of ammonia oxidation reaction (AOR) on Pt-based catalysts, revealing the key factors determining catalytic stability. In situ electrochemical surface-enhanced Raman spectroscopy (EC-SERS) detected the formation of *N3– on pure Pt catalysts and its further oxidation to toxic NOx intermediates. In stark contrast, the surface of PtIr alloy catalysts forms *N2H3 intermediates through the coupling of *NH2 and *NH species, which are gradually dehydrogenated to ultimately generate N2. Throughout the electrochemical process, no nitrogen oxides were detected on the alloy surface. The transition metal and intermediate ten-electron counting rule validated by DFT simulations theoretically explains the reason for the preferential formation of *N2H3 at the Pt-Ir synergistic catalytic sites. This key intermediate formed through the synergistic action of Pt-Ir dual sites allows the reaction to avoid pathways leading to catalyst deactivation, thereby elucidating the mechanism of enhanced durability of PtIr alloys. This study provides a theoretical blueprint for designing anti-poisoning AOR electrocatalysts through atomic-scale active site engineering.
Original link: https://doi.org/10.1021/jacs.5c09123
Editor: Fan Xiulin
Reviewer: Zhang Qin