
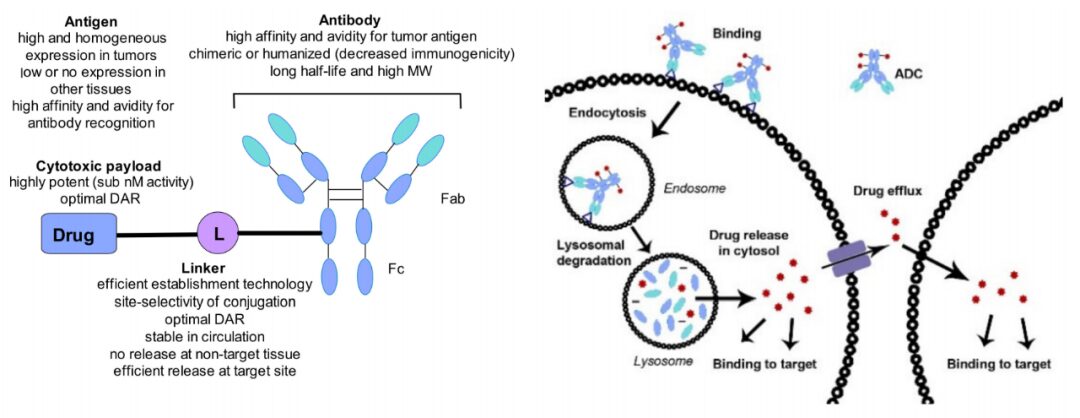
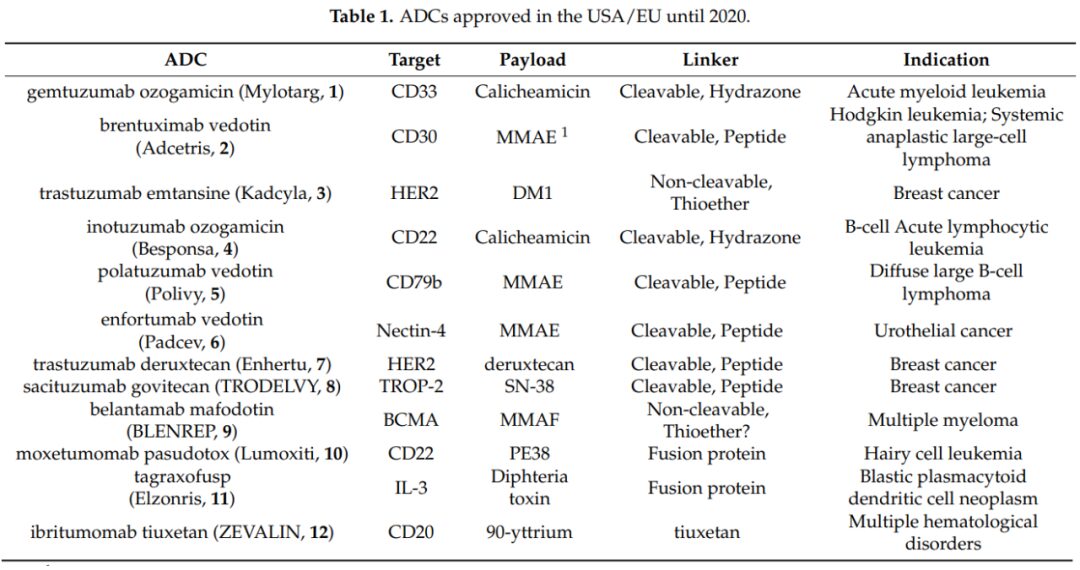
Antibody-drug conjugates (ADCs) are formed by linking monoclonal antibodies targeting specific antigens with small molecule cytotoxic drugs through linkers, combining the powerful killing effects of traditional small molecule chemotherapy with the tumor-targeting properties of antibody drugs. ADCs consist of three main components: the antibody responsible for selectively recognizing cancer cell surface antigens, the drug payload responsible for killing cancer cells, and the linker that connects the antibody and payload.


Payloads
1. Microtubule Disruptors
Monomethyl auristatin E (MMAE)


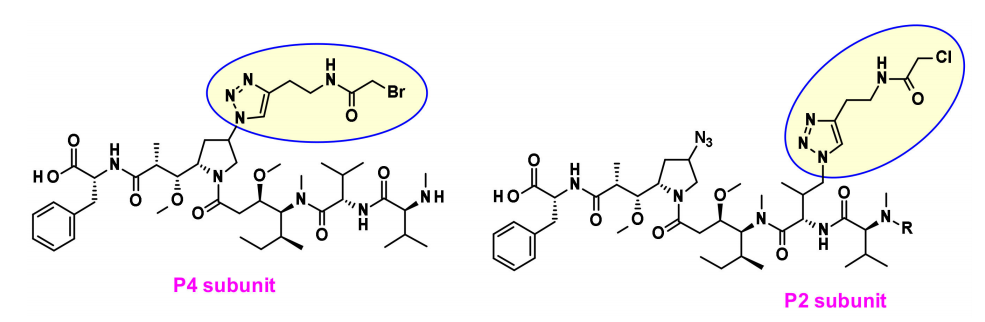
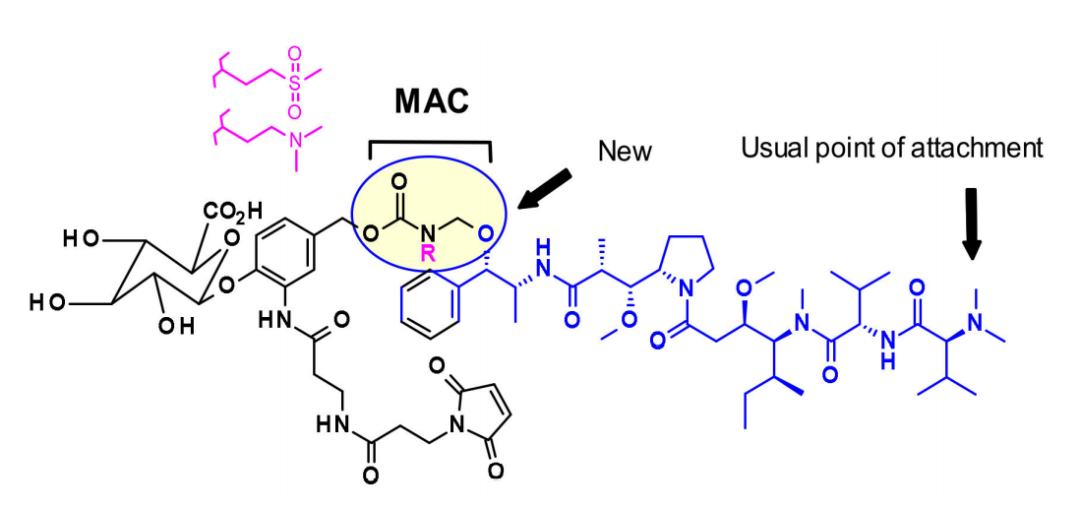

Maytansine Derivatives (DM2, DM4)

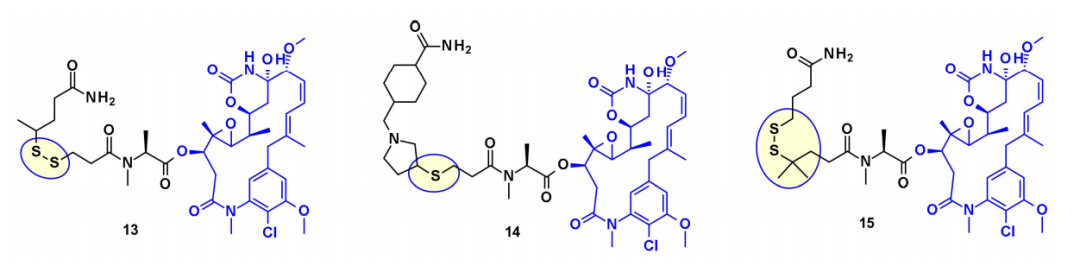
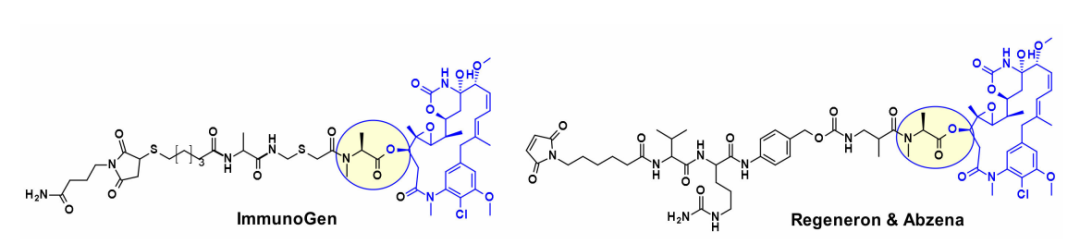
Microtubule Disruptors
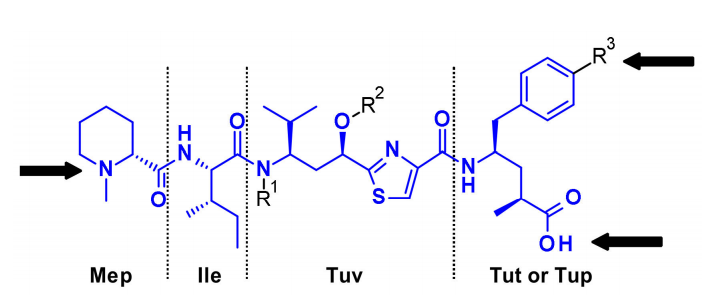

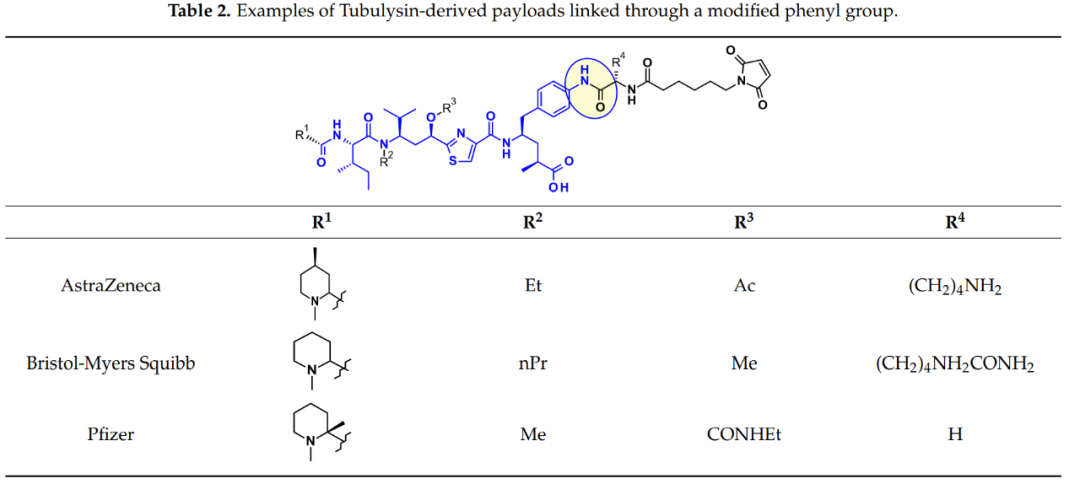
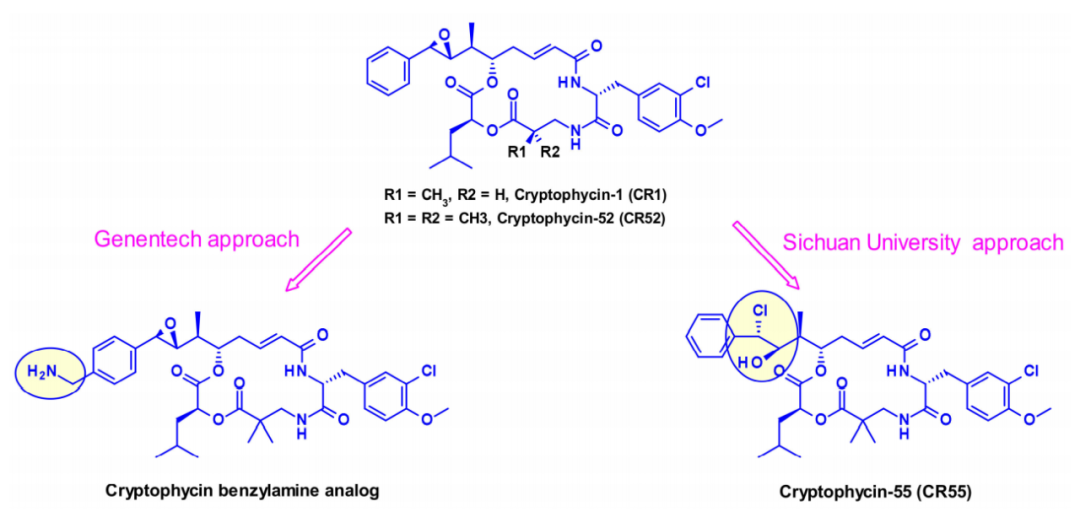
Cryptomycins
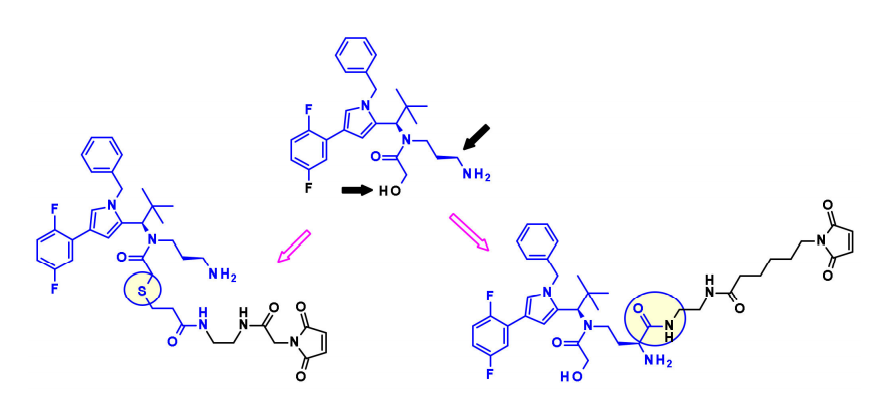
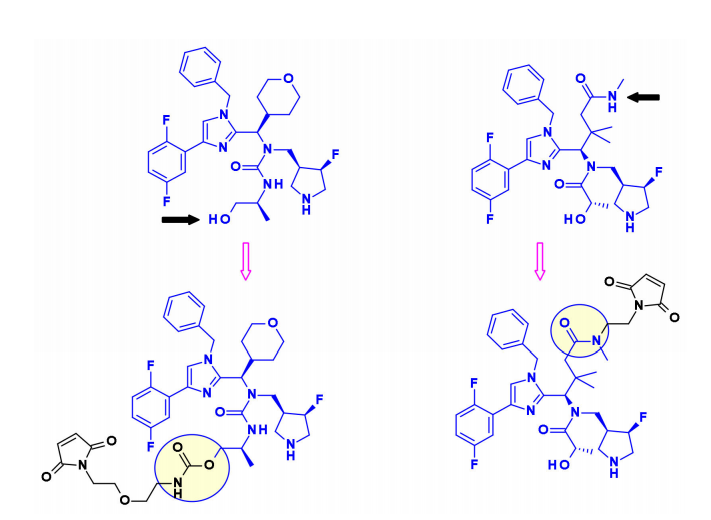
Mitotic EG5 Inhibitors
2. DNA-Damaging Drugs
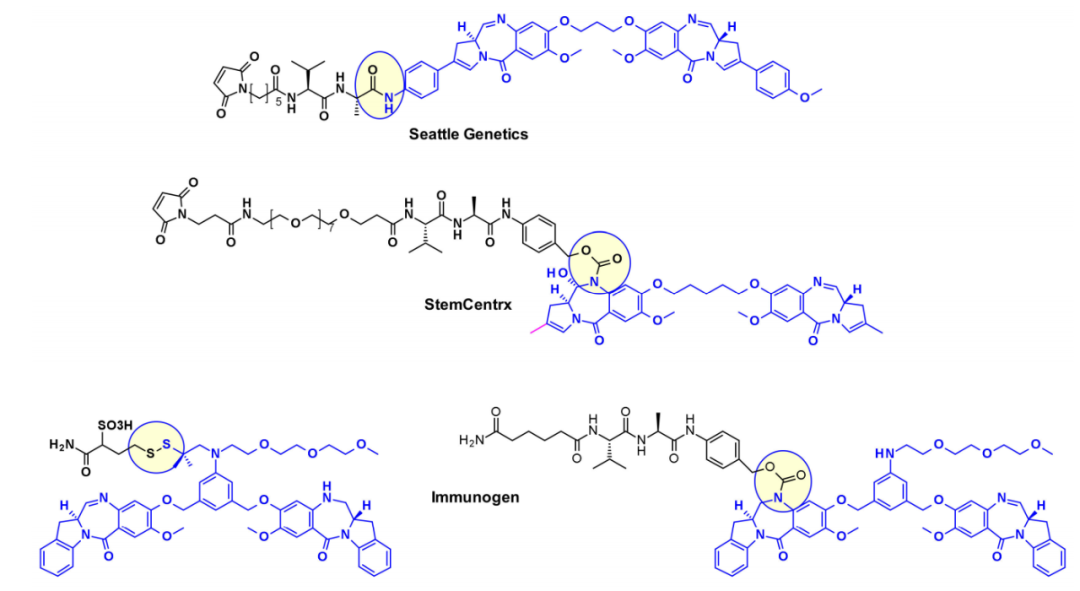
Pyrrolobenzodiazepines and Indole Chlorobenzodiazepines

Doxorubicin


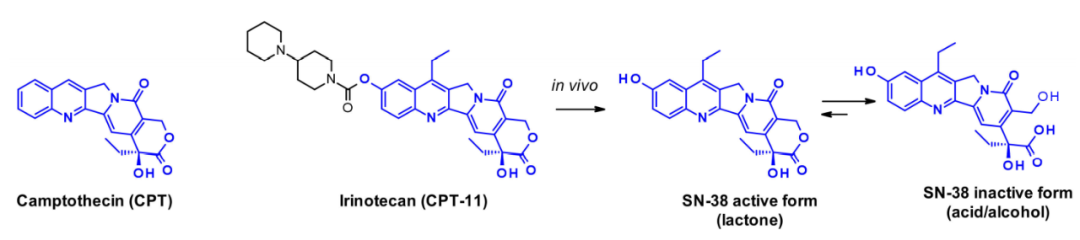
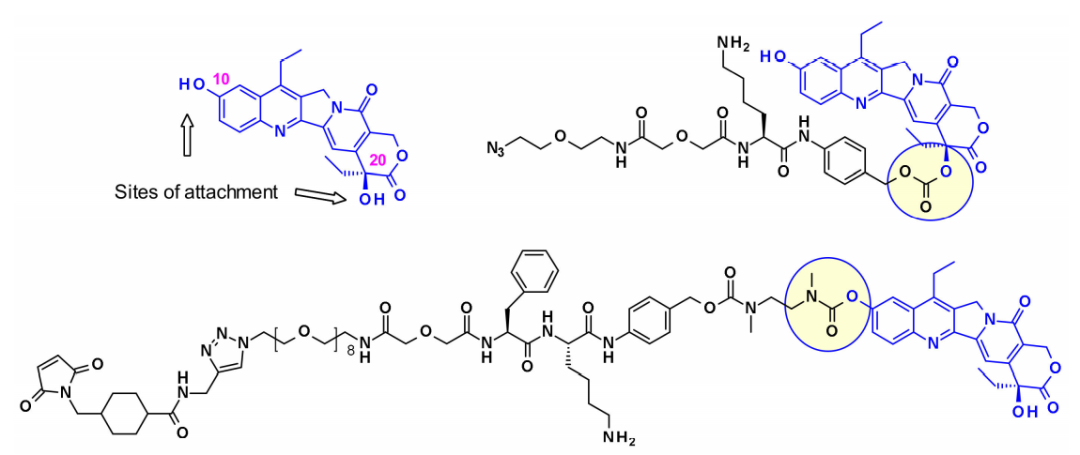
Camptothecin



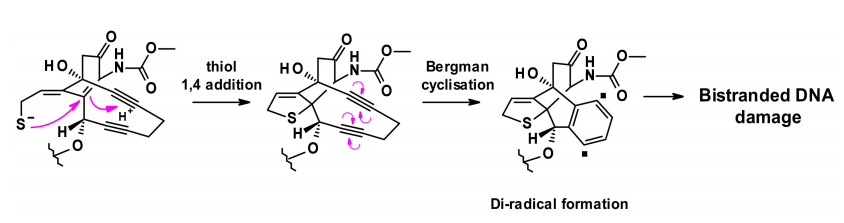
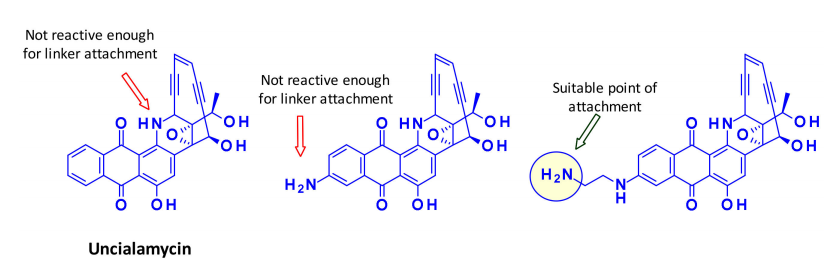
Calicheamicin



3. Innovative Drugs
Apoptosis Inducers (Bcl-xL Inhibitors)
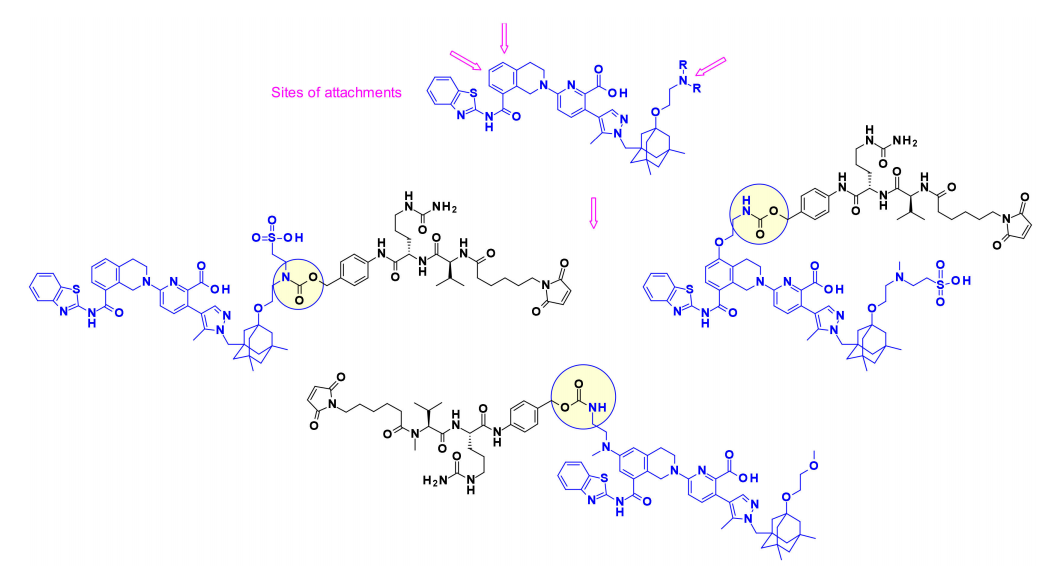
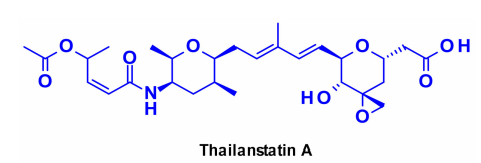
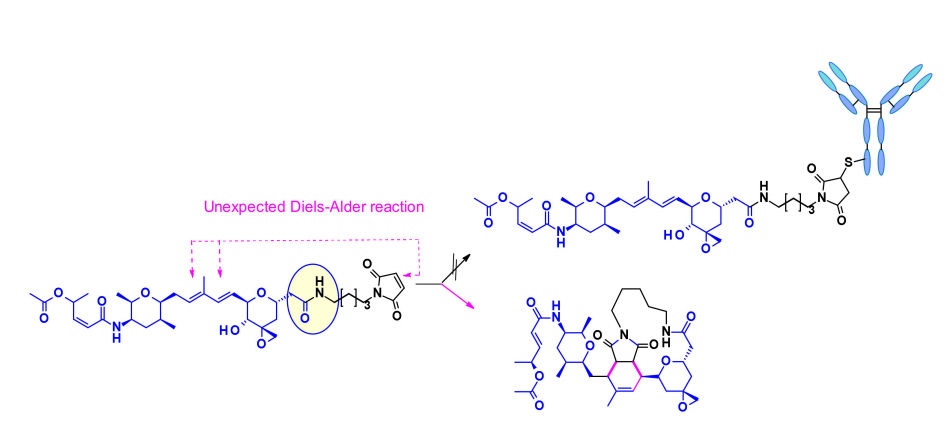
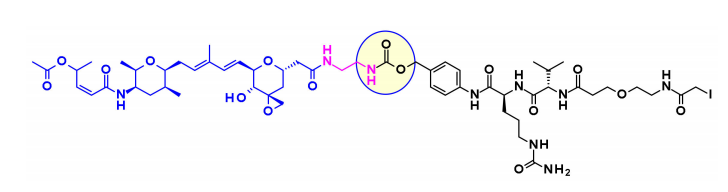
Thailanstatin A and Its Analogues

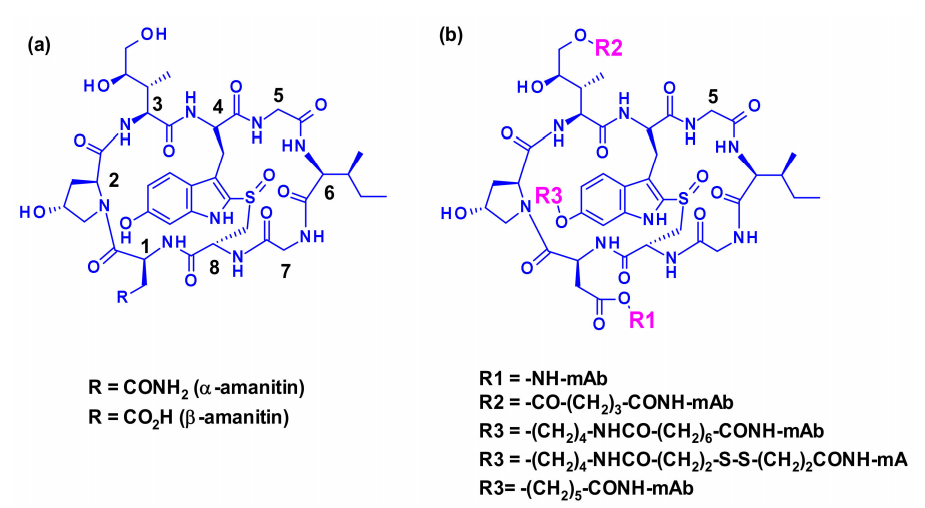
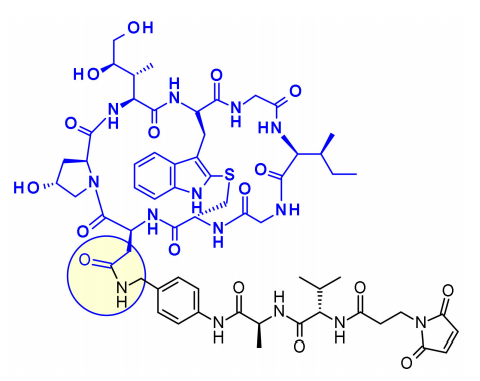
Amanitins
<img src=