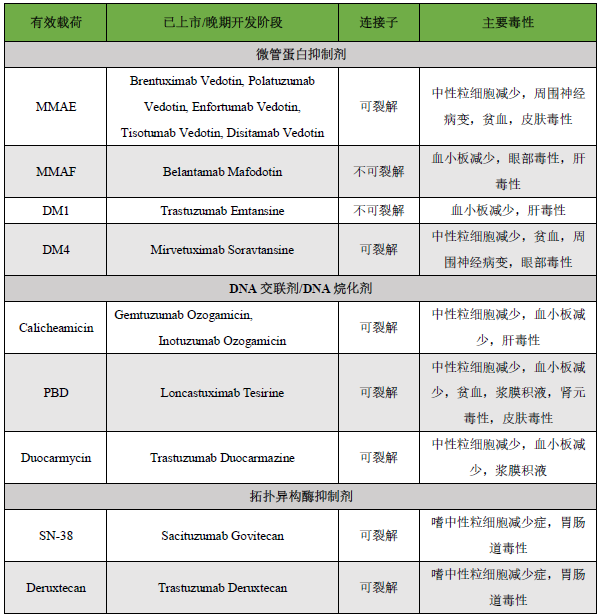
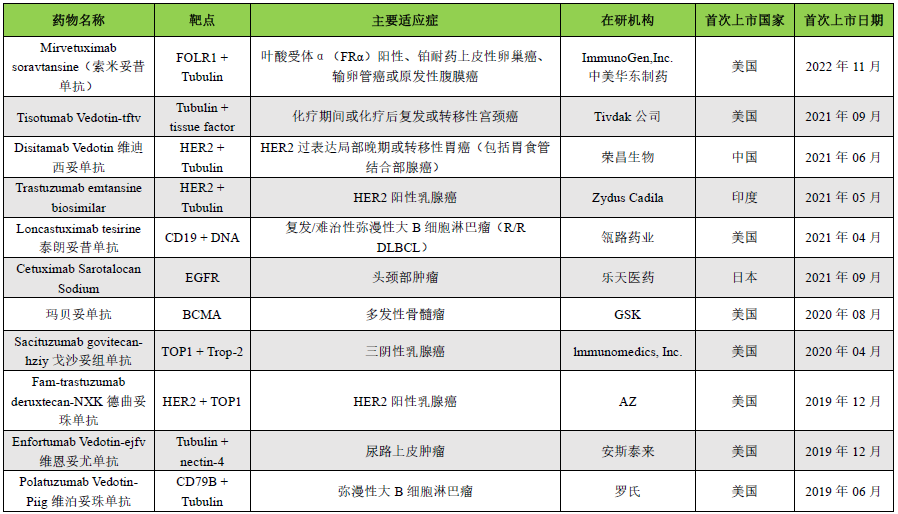
Key Content Preview:In recent years, antibody-drug conjugates (ADCs) have developed rapidly. As of August 2023, there are a total of 15 ADCs on the market globally, with over 1200 drugs under research. In June this year, Nature Biotechnology published a brief report titled “ADCs’ Revival”, highlighting the “breakthrough momentum” of ADCs in new drug development. As ADCs advance in clinical applications, some drugs have shown certain toxic side effects, leading to reduced clinical dosages or discontinuation. Some ADCs have also been terminated during the market application process due to concerns over the risk/benefit ratio or toxicity. This article summarizes the mechanisms that lead to ADC toxicity and attempts to explore strategies to enhance ADC tolerability. A summary of marketed ADCs is provided in the table below.
 In the past three years, the number of ADCs approved by the FDA has doubled. During the drug marketing phase, although some ADCs have demonstrated sufficient efficacy and safety, almost all ADCs exhibit certain toxicities during clinical application. Clinical data analysis indicates that dose-limiting toxicity (DLT) manifests similar toxicities due to the same payload. DLT is usually associated with cells and tissues that do not express the targeted antigen (i.e., off-target toxicity), limiting the ADC dosage below the optimal level for anticancer efficacy. This article reviews the mechanisms leading to ADC toxicity, summarizes common treatment-related adverse events, and discusses methods to mitigate ADC toxicity.
It is estimated that after ADCs enter the human body, only about 0.1% of the drug is delivered to the targeted tumor cell population, with the vast majority decomposing “off-site” within non-target healthy cells, potentially causing unnecessary toxicity. ADC toxicity that does not reach the target lesions or tissues can be classified as “on-target” or “off-target”, meaning that target-mediated toxicity occurs when ADC binds to antigen proteins on healthy cells.Each component of the ADC, including the antibody, linker, and payload, can influence the degree of toxicity induced by the ADC. The toxicity mechanisms of ADCs are illustrated in Figure 1.
In the past three years, the number of ADCs approved by the FDA has doubled. During the drug marketing phase, although some ADCs have demonstrated sufficient efficacy and safety, almost all ADCs exhibit certain toxicities during clinical application. Clinical data analysis indicates that dose-limiting toxicity (DLT) manifests similar toxicities due to the same payload. DLT is usually associated with cells and tissues that do not express the targeted antigen (i.e., off-target toxicity), limiting the ADC dosage below the optimal level for anticancer efficacy. This article reviews the mechanisms leading to ADC toxicity, summarizes common treatment-related adverse events, and discusses methods to mitigate ADC toxicity.
It is estimated that after ADCs enter the human body, only about 0.1% of the drug is delivered to the targeted tumor cell population, with the vast majority decomposing “off-site” within non-target healthy cells, potentially causing unnecessary toxicity. ADC toxicity that does not reach the target lesions or tissues can be classified as “on-target” or “off-target”, meaning that target-mediated toxicity occurs when ADC binds to antigen proteins on healthy cells.Each component of the ADC, including the antibody, linker, and payload, can influence the degree of toxicity induced by the ADC. The toxicity mechanisms of ADCs are illustrated in Figure 1.
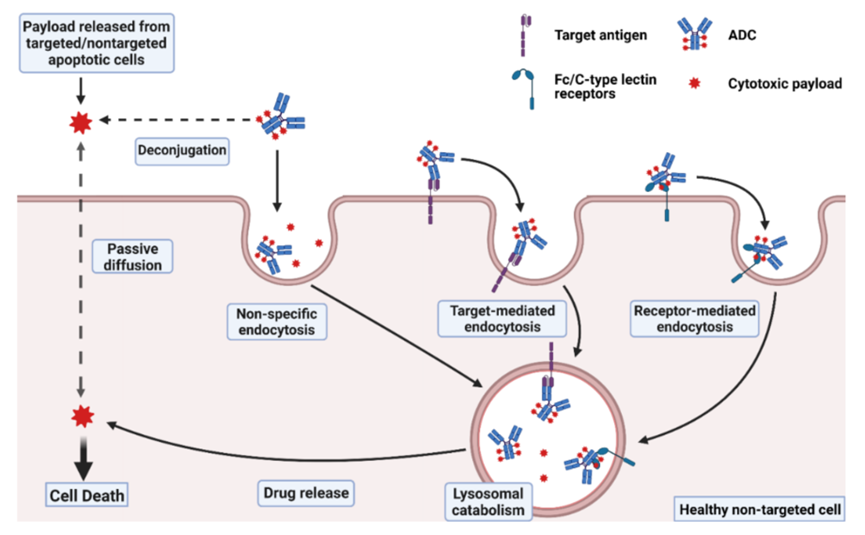
Figure 1 ADC Toxicity Mechanisms (The complete ADC may enter normal cells through non-specific endocytosis or by binding to target antigens or Fc/C-type lectin receptors for internalization. The payload released from ADC molecules in the extracellular fluid, or other targeted/non-targeted apoptotic cells, can enter normal cells through passive diffusion or non-specific endocytosis.)
“Off-Target and Off-Site” Toxicity: Theoretically, ADCs are expected to enhance therapeutic selectivity by coupling technology, directing cytotoxic payloads to target cells while minimizing payload delivery to non-target healthy tissues, thereby expanding the therapeutic index. In the early development of ADC drugs, it was believed that the toxicity of the drug was determined by the degree to which normal tissues express the target antigen or the differential expression of target antigens between tumor cells and healthy cells. However, as ADC drugs have advanced in clinical applications, dose-limiting toxicity (DLT) is rarely driven by target expression in healthy tissues. A review of ADC drug IND submission data found that ADCs composed of the same class of linkers/payloads exhibit highly similar toxicity profiles, DLT, and maximum tolerated doses (MTD). For example, ADCs containing monomethyl auristatin E (MMAE) have similar phase II clinical starting doses (1.8 mg/kg to 2.4 mg/kg). ADCs with MMAE consistently demonstrate similar DLTs (severe myelosuppression, sepsis, and severe motor neuropathy). Similarly, a review of clinical ADC data published between 2010 and 2014 found that common grade 3/4 toxicities associated with ADCs were consistent with the payload class. For instance, ADCs with MMAE payloads commonly exhibit serious anemia, neutropenia, and peripheral neuropathy. ADCs with DM1 payloads typically show grade 3/4 thrombocytopenia and hepatotoxicity, while MMAF and DM4 ADCs exhibit severe ocular toxicity. Such examples are not uncommon.
Thus, ADC “Off-Site Toxicity” is related to the off-target delivery of cytotoxic payloads, which is a key driving factor for ADC tolerability, ultimately determining the recommended dose for patients. Of course, target-mediated toxicity is not the main concern when clinically evaluating and using ADCs. Drugs with severe target-mediated toxicity can often be quickly identified in the early stages of preclinical development and ultimately discontinued.
Off-Target Delivery of ADC Payloads: After administration of ADCs, released payloads rapidly enter systemic circulation, and some payloads may be related to linker instability. Linkers mainly fall into two categories: cleavable and non-cleavable. Cleavable linkers are designed to take advantage of the unique microenvironment of tumor cells or the intracellular environment, exhibiting good stability in systemic circulation. However, in practice, it has been found that cleavable linkers can rapidly cleave in plasma, leading to premature release of payloads before reaching target tissues. Lipophilic payloads exhibit high permeability through the plasma membrane, thus effectively entering non-target cells and potentially causing cytotoxicity. For example, the hydrazone linker used in first-generation calicheamicin-based ADCs couples gemtuzumab with ozogamicin, releasing the payload in the acidic environment of the lysosome (i.e., entering the cell through receptor-mediated endocytosis). However, this linker rapidly hydrolyzes in plasma, resulting in significant premature release of payloads before the ADC contacts the target antigen, decreasing the proportion of intact ADC delivered to target cells (mechanism illustrated in Figure 2).
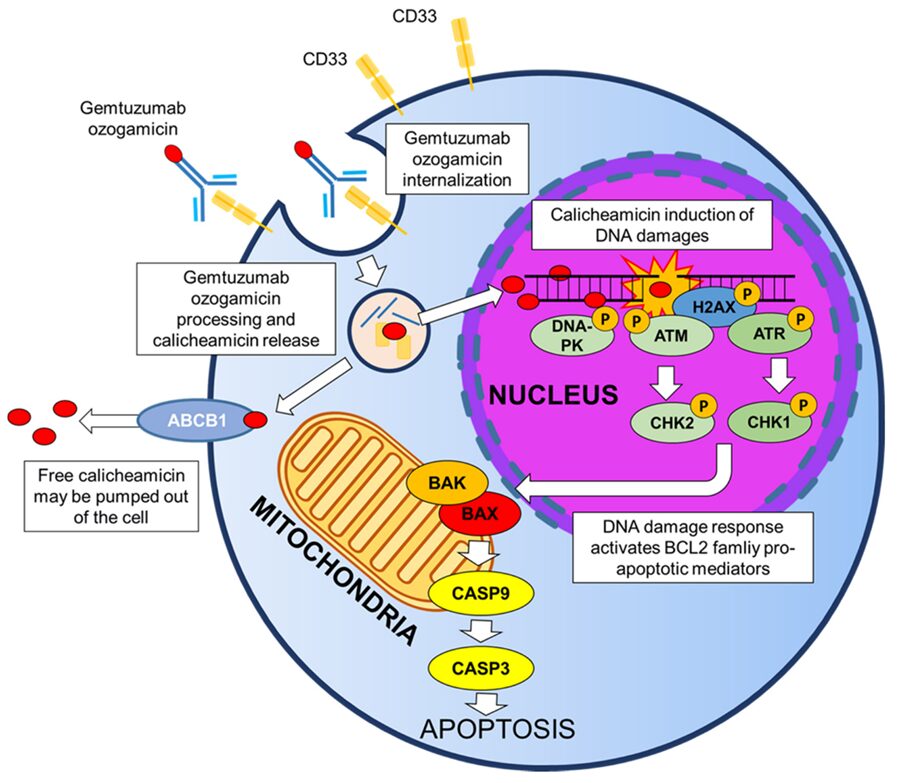
Figure 2 Mechanism of Gemtuzumab Ozogamicin
ADCs with cleavable linkers may be considered prodrugs, with 100% of the administered drug (i.e., payload) ultimately released via linker hydrolysis. In most cases, it is expected that the clearance (CL) of the payload occurs through processes like renal filtration and biliary excretion, where the elimination pathway and the efficiency of free payload CL are not influenced by the site of payload release (i.e., premature hydrolysis in plasma or within targeted or non-targeted cells post-endocytosis). Basic pharmacokinetic theory predicts that the cumulative exposure of released payloads in plasma (e.g., the area under the plasma concentration-time curve of free payload, AUC) is a simple function of the dose of the ADC or payload and the CL of the payload (i.e., AUC = Dose/CL). Therefore, poorly stable linkers are unlikely to affect the AUC of the payload in plasma. However, poorly stable linkers reduce the exposure ratio of the payload at the target site relative to plasma, thus decreasing the ratio of on-site to off-site ADC cytotoxicity (relative to toxicity reducing efficacy). As linker technology advances, the stability of ADCs has gradually improved (relative to first-generation ADCs). However, due to the plasma protein sensitivity of peptide-based linkers, linkers containing disulfide bonds or maleimide groups can react with active thiols in plasma, and alkyl carbamate esters are sensitive to serum esterases, leading to non-selective and premature release of payloads in systemic circulation.
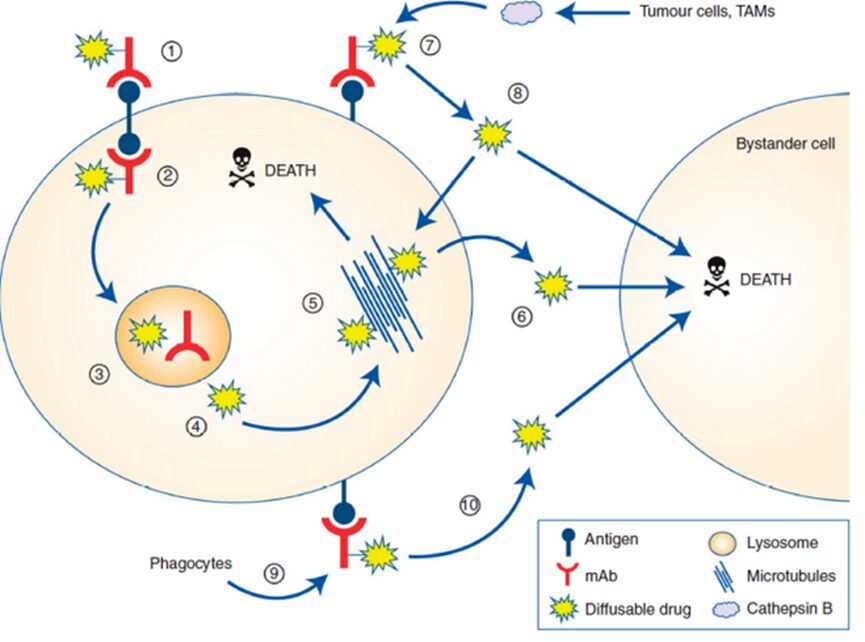
Compared to cleavable linkers, non-cleavable linkers are more stable in plasma but require the ADC to enter cells, where they dissociate and metabolize through proteolysis. These metabolites usually consist of complete linker payloads linked to antibody-conjugated amino acid residues, such as lysine-SMCC-DM1 from trastuzumab-maytansine conjugates or cysteine-MC-MMAF from belantamab mafodotin. These metabolites are usually charged and have poor cell membrane permeability. ADCs using non-cleavable linkers exhibit higher tolerability, which may be related to reduced off-target toxicity of released payloads, but ADCs with cleavable linkers typically show better efficacy, possibly due to the bystander effect ((bystander cell)), where surrounding cells (Figure3) including heterogeneous tumor cells and the aforementioned metastatic cells, can be either Ag+ or Ag-. The bystander effect allows the drug to also kill these cells.
Figure 3 Schematic Diagram of Bystander Effect of ADCs
ADCs with lipophilic payloads and cleavable linkers account for over 80% of the currently approved ADCs, making them the preferred choice for treating tumors associated with heterogeneous antigen expression or slower antigen/ADC internalization rates. In addition to enhancing antitumor potency, the bystander effect also exacerbates off-target toxicity of ADCs due to the increased distribution of lipophilic membrane-permeable payloads in normal tissues.
Besides the passive diffusion of released payloads into non-target cells through the plasma membrane, the non-specific endocytosis of intact ADCs may also contribute to the off-site delivery of payloads. Non-specific endocytosis may be influenced by the hydrophobicity and charge of the ADC. Since most drug linkers used in ADC platforms are highly lipophilic, the hydrophobicity of ADCs is usually proportional to the drug load (i.e., drug-antibody ratio, DAR). In a study by Hamblett et al., several anti-CD30-vc-MMAE ADCs with DAR values of 2, 4, and 8 were evaluated for pharmacokinetics, efficacy, and toxicity in vivo. ADCs with higher DAR values exhibited faster systemic clearance rates, lower tolerability, and narrower therapeutic indices compared to ADCs with lower DAR values. Similarly, Sun et al. demonstrated that ADCs with a DAR of 10 had a clearance rate five times higher than those with a DAR below 6, with reduced efficacy and tolerability in vivo. Mice treated with low DAR ADCs (2 and 3.5) showed less weight loss (around 4%) compared to those treated with high DAR ADCs (weight loss of 7-9%). Additionally, the distribution of ADCs with higher DAR in the liver was significantly increased, possibly due to non-specific uptake by Kupffer cells and hepatic sinusoidal endothelial cells.
Positively charged molecules typically increase charge-mediated endocytosis due to ionic attraction to negatively charged cell membranes. Several preclinical studies have shown that the plasma clearance and tissue distribution of IgG antibodies correlate with their isoelectric points (pI). For example, some studies have shown that positively charged antibodies exhibit higher uptake in the liver and spleen of mice compared to neutral or negatively charged antibodies, resulting in reduced plasma exposure.
Off-Target Receptor-Mediated Uptake of ADCs: As part of the immune system, IgGs interact with receptors that express Fc on the surface of immune cells through the constant (Fc) domain, signaling with different types of immune cells. One of the main types of Fc receptors interacting with IgG is the Fc gamma receptor (FcγR). Binding to the Fc domain may lead to non-target-dependent uptake and toxicity of ADCs in immune cells. Studies have indicated that FcγRs may be associated with thrombocytopenia related to trastuzumab emtansine (T-DM1) therapy. Thrombocytopenia is considered an off-target dependent toxicity since platelets and megakaryocytes (MKs) do not express the target HER-2 of T-DM1. T-DM1 minimally affects mature MKs while being internalized, exhibiting strong cytotoxicity during the differentiation of MKs from human bone marrow. Off-target toxicity appears to be mediated by FcγRIIa, and blocking FcγRIIa binding can inhibit the uptake of T-DM1. Interstitial lung disease (ILD)/pneumonia is one of the common life-threatening adverse events associated with anti-HER2 ADC therapy. However, the potential causes of ADC-induced ILD remain unclear. In a recent study, Kumagai et al. confirmed a higher distribution of trastuzumab in alveolar macrophages, given that alveolar macrophages express excess FcγR and low HER2, Fc-mediated non-specific uptake may contribute to ADC-induced ILD. Research on ADCs that remove FcγR binding may help determine the role of FcγR in ADC-induced ILD.
In addition to FcγRs, mannose receptors (MR) binding and receptor-mediated endocytosis are also potential mechanisms mediating ADC off-target hepatotoxicity. Receptor-mediated MR uptake is one of the key clearance mechanisms. For instance, in humans, therapeutic IgGs with high levels of mannosylation exhibit significantly faster systemic clearance rates than normal IgGs. Kogelbegs et al. confirmed MR-mediated uptake of mannosylated antibody-enzyme fusion protein MFECP1 by hepatic sinusoidal endothelial cells (SECs). The MR inhibitor mannan significantly inhibited MFECP1 clearance. Furthermore, acute thrombocytopenia and sinusoidal obstruction syndrome (SOS) associated with ADC treatment are related to the degeneration and loss of hepatic SECs. Therefore, MR-mediated uptake by hepatic SECs may be a major cause of non-target-dependent hepatotoxicity of ADCs.
“Off-Site and On-Target” Toxicity: Although evidence suggests that payload-mediated off-target mechanisms lead to most ADC toxicity, the binding of ADCs to target antigens expressed in healthy tissues may also lead to significant toxicity. For instance, 40% of patients experience taste disturbances when using enfortumab vedotin, which is due to the expression of the ADC target (nectin-4) in salivary glands. In fact, this toxicity is not commonly seen in patients treated with other approved MMAE-ADCs. As shown in the above example, the toxicity produced by ADCs is unrelated to the payload. Conversely, ADCs constructed with different payloads exhibit the same toxicity, suggesting that this toxicity is target-mediated.
It is noteworthy that applying the same ADC to treat different tumors may lead to different toxicities. For example, severe rash is one of the dose-limiting toxicities of Glembatumumab vedotin (CDX-011), which may be related to the expression of the target gpNMB in the skin. After administering the same dose of CDX-011, 4% of patients with advanced breast cancer experienced ≥3 grade rashes, while the incidence was over 30% in patients with advanced multiple myeloma. The incidence of itching and hair loss in melanoma patients is also higher than that in breast cancer patients (63% and 65% versus 21% and 25%). The mechanism behind this phenomenon is unclear but may relate to the expression of gpNMB in healthy cells of these two tumors. In another example, LOP628, an anti-KIT-SMCC-DM1 ADC, caused life-threatening rapid hypersensitivity reactions (HSR) in some patients, leading to the termination of clinical development. The primary cause of HSR is due to the co-participation of the anti-KIT monoclonal antibody in KIT and FcγRs, causing degranulation of mast cells. This unique situation presents a mechanism where target-mediated toxicity does not involve toxicity related to antigen-expressing cells but acts through co-stimulatory signaling pathways to activate the immune system. Furthermore, the expression of target cell antigens in healthy tissues does not always lead to target cell toxicity. For instance, membrane-associated mucin MUC16 is expressed in the epithelial cells of the human eye; however, patients treated with anti-MUC16 MMAE-ADCs in clinical trials did not experience ocular toxicity. TROP-2 is widely expressed in various tissues; however, sacituzumab-govitecan (gogotuzumab), an ADC conjugated with anti-TROP2 SN-38, exhibits clinical toxicity characteristics similar to those of the SN-38 payload, suggesting that its toxicity mechanism may be off-target. The low target toxicity of gogotuzumab may relate to: (1) limited expression of TROP2 in non-malignant tissues compared to tumors; (2) insufficient expression to induce a toxic response; (3) lower sensitivity of normal tissues to the SN-38 payload compared to tumor tissues. Additionally, due to the use of pH-sensitive linkers in gogotuzumab, payload release occurs in an acidic environment, which may also be one of the reasons for its low target-mediated toxicity compared to tumors.
Clinical Toxicity Analysis of ADCs
ADC-Related Dose-Limiting Toxicities
Neutropenia: Neutrophils constitute 40% to 70% of granulocytes and are an important component of the innate immune system, rapidly produced in the bone marrow through differentiation from hematopoietic stem cells (about 1011 cells daily). Their circulating half-life is relatively short (about one day). Compared to other long-lived bone marrow cells, including platelets (8 days) and red blood cells (120 days), these unique characteristics make neutrophils more susceptible to antitumor drugs. Disruption of hematopoietic cell division in the bone marrow often leads to neutropenia and increases the risk of severe infections. Severe neutropenia is a common dose-limiting toxicity associated with most MMAE-ADCs utilizing valine-citrulline cleavable linkers, including brentuximab vedotin, polatuzumab vedotin (Polivy®), enfortumab vedotin (PADCEV), and tisotumab vedotin (Tivdak™).
ADC-Related Neutropenia is associated with cumulative plasma exposure to released payloads. Payloads with cell membrane permeability easily distribute to the bone marrow and differentiating hematopoietic cells. The dissociation of intact ADCs in the extracellular fluid of the bone marrow may also lead to myelosuppression, as differentiated neutrophils secrete serine proteases that can cleave ADC linkers. Besides the aforementioned off-target mechanisms, target-dependent mechanisms may also contribute to neutropenia in ADCs targeting leukemia antigens. In a recent systematic analysis, Haubner et al. quantified the expression of leukemia stem cells. Some of these genes, including CD33, CD123, and CLL-1, are highly expressed not only in leukemia stem cells but also in normal hematopoietic stem/progenitor cells, where CD33, CD123, and CLL-1 are precursors for neutrophil production.
Thrombocytopenia: Thrombocytopenia is a common off-target toxicity associated with ADCs utilizing stable linkers (SMCC-DM1 or mc-MMAF) and DNA crosslinking payloads like calicheamicin. For instance, 99% of acute myeloid leukemia patients and 42% of acute lymphoblastic leukemia patients receiving gemtuzumab ozogamicin experienced ≥3 grade thrombocytopenia. Reportedly, 14.5% of breast cancer patients treated with trastuzumab emtansine and 21% of multiple myeloma patients treated with belantamab mafodotin experienced ≥3 grade thrombocytopenia.
The exact mechanism of thrombocytopenia is unclear. One potential cause may be the non-specific uptake of ADCs through FcγR-mediated or micropinocytosis pathways during MK differentiation. However, the clinical manifestation of ADC-related thrombocytopenia typically occurs within 24 hours, significantly shorter than the lifespan of platelets (8-10 days). In fact, Guffroy et al. demonstrated that ADCs based on calicheamicin showed damage to sinusoidal epithelial cells after 3 days of administration in crab-eating macaques, with platelet accumulation in hepatic sinusoids correlating with serum thrombocytopenia.
Peripheral Neuropathy: Clinical manifestations of peripheral neuropathy include sensory-related symptoms such as numbness, tingling, and pain in the limbs, or to a lesser extent, motor-related symptoms like muscle weakness. Peripheral neuropathy is a common toxic side effect associated with chemotherapy drugs that inhibit microtubules. Similarly, peripheral neuropathy is a dose-limiting off-target toxicity associated with ADCs utilizing microtubule-inhibiting payloads connected via cleavable linkers, such as vc-MMAE, SPP-DM1, and SPDB-DM4. The mechanism of peripheral neuropathy is related to the release of free payloads into systemic circulation causing axonal peripheral nerve damage. Although peripheral neurons do not proliferate significantly, functional microtubules are critical for transporting proteins from the cell body to distal synapses. Therefore, microtubule disruption by microtubule-inhibiting payloads may lead to peripheral neuropathy.
Ocular Toxicity: Ocular toxicity is one of the key off-target dose-limiting toxicities of ADCs containing the SPDB-DM4 linker-payload, such as cantuzumab ravtansine, mirvetuximab soravtansine, coltuximab ravtansine, or ADCs containing mc-MMAF, such as belantamab mafodotin, AGS-16C3F, SGN-75. The susceptibility of the eye to ADC cytotoxicity may relate to the ample blood supply to the eye, the presence of rapidly dividing epithelial cell populations, and high expression of targets in the eye. In clinical applications, the symptoms of ADC-related ocular adverse events include blurred vision, keratitis, dry eye, and microcystic epithelial damage. The mechanism of ocular toxicity related to ADCs is currently unclear. Some experts also believe that non-specific uptake of intact ADCs via macropinocytosis may be a mechanism for off-target ocular toxicity. The table below summarizes common dose-limiting toxicities associated with ADC treatment.
Table 1 Dose-Limiting Toxicities Related to Payloads
Strategies to Reduce Toxicity
High loss rates of ADCs containing mitotic inhibitory payloads are a major cause of toxic side effects in clinical trials. Often, increasing the dose improves efficacy while also increasing toxic side effects. However, raising the dose increases the incidence of life-threatening events, thereby limiting clinical development. For example, vadastuximab talirine, a CD33 PBD-ADC, achieved a 70% complete remission rate in AML patients; however, clinical development was terminated due to some treatment-related fatal events. An analysis by the FDA found that from 2013 to 2017, 47% of new drug applications for PBD-conjugated ADCs were halted primarily due to safety concerns. These results indicate that due to the toxic side effects of ADCs, their clinical application doses fall below effective doses, thus limiting clinical development.
Payload/Linker Coupling Technology: Using traditional non-specific coupling methods with amino lysine residues or thiol cysteine residues leads to significant heterogeneity in DAR and hydrophobicity. The drug load ranges from no load (DAR = 0) to high-load ADCs (DAR ≥ 6). High-load ADCs are unstable in plasma and exhibit higher non-specific uptake rates in the liver, leading to increased off-target toxicity. With technological iterations, ADCs with closer DAR values have been developed. The first site-specific coupling strategy was developed by Junutula et al., which involved site-specific coupling of the payload to the cysteine residue at position Ala114 of the antibody CH1 domain. By genetically engineering the insertion of cysteine residues at specific points in the antibody, the thiol (-SH) of the cysteine can be coupled with the toxin to form site-specific antibody-drug conjugates. This technology is known as Thiomab site-specific coupling.
In addition to site-specific coupling, modified linkers can also alter the hydrophobicity of ADCs, reducing toxicity. Linker PEGylation increases the hydrophilicity of ADCs, improving PK, tolerability, and efficacy. The development of self-cleaving linkers has also enhanced the stability and tolerability of ADCs. In the early 1980s, Carl et al. developed para-aminobenzyloxycarbonyl (PABC) self-cleaving linkers for prodrug design. Since then, PABC linkers have been widely used in ADCs, particularly those conjugated with MMAE. After ADC internalization into target cells, they are cleaved by lysosomal enzymes, and the PABC spacer in the vc-PAB-MMAE linker/payload undergoes a cascade decomposition reaction, promoting the release of MMAE intracellularly. Compared to earlier cleavable linkers, the vc-PABC linker demonstrates superior plasma stability and better toxicity profiles, as seen in pH-sensitive linkers used in calicheamicin-based ADCs or disulfide bond-containing linkers used in DM1-based ADCs, which have achieved success in 4 of the 12 ADCs approved by the FDA based on MMAE. Additionally, PABC linkers have also been utilized in the development of PBD-based ADCs. Recently,Pillow et al. developed complex self-cleaving linkers that enable cytotoxic payloads to be directly coupled to engineered cysteine residues (Figure4). Engineered cysteine coupling allows for uniform distribution of cytotoxic loads across the entire ADC. IgG antibodies contain eight cysteine bonds, enabling the engineering of ADC drugs with DAR values of 2-4. Companies like Pfizer and others have adopted this technology as a method for ADC drug coupling. These ADCs show tremendous potential and tolerability in preclinical phases compared to traditional ADCs. Compared to traditional peptide-PABC linker technology, this technology has improved the tolerability of PBD-ADCs. Traditional peptide-ADCs and self-cleaving disulfide-ADCs exhibit similar in vivo efficacy. However, regarding toxic side effects, self-cleaving ADCs in rats can reach MTDs of up to 10 mg/kg, while animals treated with peptide-ADCs at a dose of 5 mg/kg experienced significant weight loss.
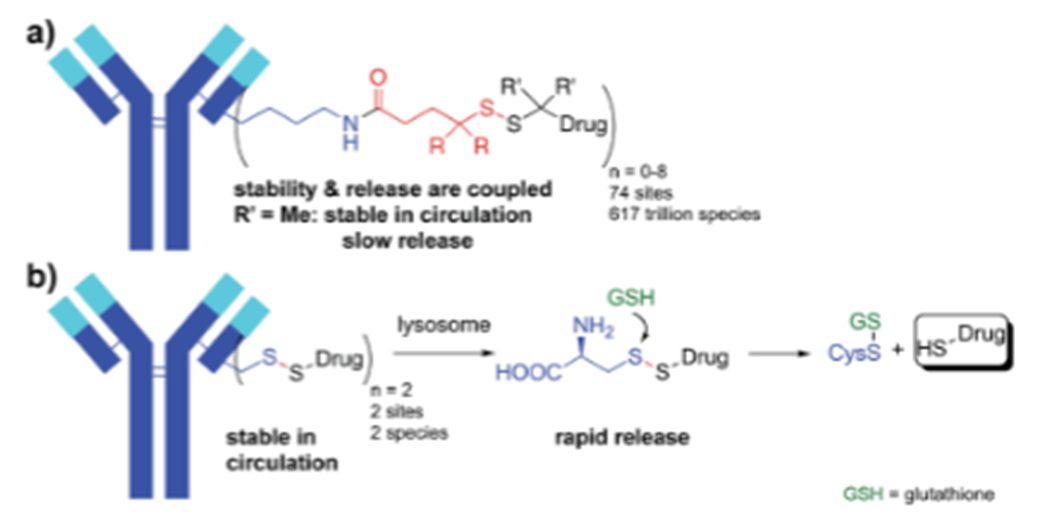
Figure 4 ADCs with Self-Cleaving Linkers Containing S-S Bonds
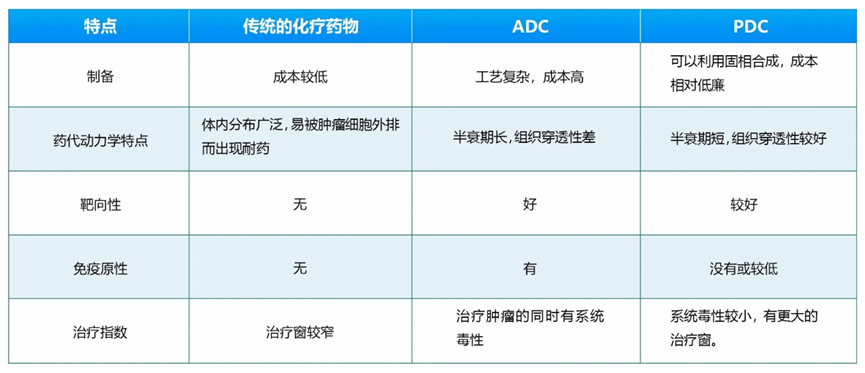
Antibody Modification: Since most ADCs have their targets expressed to some extent in healthy tissues, the delivery of cytotoxic payloads by ADCs in these tissues can lead to “on-target off-site” toxicity. To reduce on-target toxicity, peptide-drug conjugates (PDC) have been developed, which are analogous to ADCs, but PDCs somewhat overcome some defects of ADCs. Compared to ADC drugs, PDC drugs have a smaller molecular weight, better tissue penetration, and lower immunogenicity; additionally, PDCs are easier to synthesize, purify, and characterize than the complex production processes of antibodies, resulting in lower costs. In recent years, the application of cyclization technology, phage display technology, and mRNA display technology for screening targeted peptides has accelerated the development of PDC drugs. PDCs are expected to become the next generation of targeted drugs following small molecule targeted drugs, antibody drugs, and ADCs. A comparison of PDCs with traditional chemotherapy drugs and ADCs is shown in the table below.
Table 2 Comparison of PDCs with Traditional Chemotherapy Drugs and ADCs
Another method to reduce on-target toxicity is through the development of bispecific antibodies. This approach enhances selectivity, as ADC binding is only observed in cells co-expressing both antigens. Studies have shown that bispecific antibodies targeting HER2+/EGFR- or HER2-/EGFR+ cells demonstrate significantly improved in vitro binding selectivity to HER2+/EGFR+ cells.
Optimizing Administration Regimens: Gemtuzumab ozogamicin was initially approved in 2001 for the treatment of CD33-positive AML at a dose of 9 mg/m2 at least every two weeks. However, severe toxic reactions were observed at this dosing frequency and level, including thrombocytopenia (99%), neutropenia (97%), ≥3 grade hyperbilirubinemia (23%), and ≥3 grade elevations in alanine or aspartate aminotransferase levels, with no significant improvement in efficacy. Consequently, gemtuzumab ozogamicin was withdrawn from the market in 2010. Recently, a graded administration of ADCs has been introduced, where patients receive a dose of 9 mg/m2 on days 1, 4, and 7 of the dosing cycle, exceeding three doses of 3 mg/m2, resulting in fewer toxic reactions. PK/PD analyses of gemtuzumab ozogamicin indicate that its toxicity is driven by peak plasma concentrations (Cmax), while its efficacy is driven by exposure (AUC).
The essence of ADC drug design is to utilize the high selective targeting and transport capabilities of antibodies to effectively deliver biologically active small molecule drugs, such as cytotoxic small molecule drugs, to the lesions, serving both as a novel drug molecule and a targeted delivery system. Within this system, key elements such as antibodies, linkers, small molecules, and coupling technologies need to be considered as a whole. Selecting suitable components and technologies based on the biological characteristics of the target and the nature and mechanisms of the disease is crucial for the successful development of ADC drugs. To further explore the potential of ADC drugs, extensive research is ongoing both domestically and internationally on various key elements to continuously enrich the toolkit for developing the next generation of ADC drugs and provide a steady stream of new ideas. These new ideas include: selecting antibodies targeting the tumor microenvironment and combining them with linker drugs that utilize extracellular release mechanisms to address target-related resistance; using smaller molecular weight antibody fragments or nanobodies to develop ADCs to improve penetration into brain tumor tissues; selecting linkers with better hydrophilicity to improve the drug load and physicochemical properties of ADCs; using small molecule drugs with different mechanisms of action to reduce side effects and expand the application fields of ADC drugs; and employing site-specific coupling technologies to improve drug load uniformity and pharmacokinetic properties. It is believed that with the joint efforts of researchers in the ADC field, overcoming the existing shortcomings and challenges of ADCs, the next generation of ADC drugs will bring new surprises to targeted therapy for tumors, benefiting more cancer patients. Furthermore, as a delivery system, ADC drugs are expected to be used in a wider range of fields in the future, such as central nervous system diseases, genetic diseases, and infectious diseases, serving as truly efficient “magic bullets” to eliminate diseases and save lives.
Pharmaceutical Research Report Sharing Camp
Collecting and sharing mainstream pharmaceutical research reports from the entire market

During the event: 66 yuan/year, only 0.2 yuan/day
Enjoy over 1400 selected pharmaceutical research reports
In the next year, an additional 1000 reports
References:
D. T N,M. B B,P. J B. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability[J]. Cancers,2023,15(3).
Michele G,Giorgia S,Alessandra S, et al. Therapeutic Targeting of Acute Myeloid Leukemia by Gemtuzumab Ozogamicin[J]. Cancers,2021,13(18).
H A S,P M B. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required?[J]. British journal of cancer,2017,117(12).
Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582.
Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804.
Introduction to peptide-drug conjugates (PDC) and their pharmacokinetic research strategies, WuXi AppTec
Liu Wenchao, Li Hongfeng, Hu Zhaohong. Technical status and prospects of antibody-drug conjugates[J]. Progress in Biochemistry and Biophysics, 2023, 50(05):1167-1189.DOI:10.16476/j.pibb.2023.0141.