


Introduction
Antibody-drug conjugates (ADC) combine the high specificity of monoclonal antibodies with the high potency of small molecule cytotoxic drugs to enhance the targeting of cancer therapies and reduce side effects. Compared to traditional antibodies or antibody fragments, ADCs can release highly active cytotoxins within tumor tissues, theoretically leading to higher efficacy. ADCs have undergone three developmental stages:
First Generation Antibody Conjugation Technology
The concept of combining the specificity of antibodies with the toxicity of drugs to create a higher level of targeted therapy can be traced back to 1913, when Paul Ehrlich proposed the development of a “magic bullet” for the selective targeting of tumors. Despite the simplicity and novelty of this concept, the first ADC was not used clinically until 2000, when the FDA approved Gemtuzumab ozogamicin (Mylotarg, Pfizer/Wyeth) for the treatment of acute myeloid leukemia. In 2011, Brentuximab vedotin (Adcetris, Seattle Genetics) was approved for the treatment of non-Hodgkin lymphoma. Two years later, Trastuzumab emtansine (Kadcyla, Genentech/Roche) was approved for the treatment of HER2-positive breast cancer. To date, 14 ADCs have been approved, including Inotuzumab ozogamicin; Polatuzumab vedotin; Enfortumab vedotin; Trastuzumab deruxtecan; Sacituzumab govitecan; Belantamab mafodotin; Moxetumomab pasudotox; and Loncastuximab tesirine.
Second Generation Antibody Conjugation Technology
2.1 Additional Introduction of Cysteine
The first systematic proof that site-specific conjugates have better efficacy came from Genentech’s Junutula and colleagues, who utilized a technology called THIOMAB site-specific conjugation technology. This technology primarily introduces an additional cysteine, which, under reducing conditions, allows the introduced cysteine and interchain disulfides to be reduced, oxidized under the action of CuSO4, and reformed disulfides, while the drug is ultimately conjugated to the additional introduced cysteine through maleimide chemistry. Compared to the first-generation conjugates with an average DAR of 3, the DAR of THIOMAB site-specific conjugation technology ADCs is 2. Researchers utilized this technology to couple MMAE with antibodies against MUC16, achieving better efficacy in ovarian cancer animal models. More importantly, the drug exhibited higher dose tolerance while improving stability in rat and monkey serum.
THIOMAB™ Site-Specific Conjugation Platform
2.2 Introduction of Non-Natural Amino Acids
Due to the growth of the ADC market and the advantages of site-specific conjugation technology, more and more researchers are committed to developing ADC drugs using second-generation antibody conjugation technology. Generally, second-generation antibody conjugation technology requires the introduction of a unique reactive group at a site-specific location in the antibody, followed by selective conjugation of a functional molecule to this unique group using bioorthogonal reactions. Currently, genetic code expansion, protein tagging, and enzyme treatments are being used to introduce unique reactive groups into proteins. In both prokaryotic and eukaryotic cells, strategies to introduce non-canonical amino acids (ncAAs) into proteins provide a relatively simple method for site-specific conjugation. A bioorthogonal ncAA—p-acetylphenylalanine (pAcF)—was first used to explore site-specific conjugation. Researchers used an acyl-tRNA synthetase (aaRS)/tRNA pair that is orthogonal to the host’s endogenous translation machinery to site-specifically insert pAcF at amber codons in trastuzumab. Using this method, researchers obtained trastuzumab containing two pAcFs (one on each heavy chain). They then coupled it with an alkoxy amine-derivatized drug monomethyl auristatin, ultimately resulting in an ADC drug with a DAR of 2, which exhibited the same efficacy as first-generation ADCs, while its safety improved to some extent.
pAcF Site-Specific Conjugation
2.3 Introduction of Specific Tags
While site-specific conjugation can be achieved through the introduction of non-natural amino acids, in production, non-natural amino acids largely limit the expression of antibodies. To avoid this issue, researchers have introduced short peptides and utilized enzyme-catalyzed reactions for site-specific conjugation. The aldehyde-tagged SMARTag short peptide sequence is CXPXR, which can be site-specifically inserted into a specific location of the antibody via genetic engineering. The cysteine in the tag can be oxidized to form formylglycine under the action of formylglycine-generating enzyme (FGE). Scientists at Redwood Bioscience found that antibodies containing formylglycine sites can utilize bioorthogonal reactions for site-specific conjugation; oxime ligation and alkoxy amines and hydrazine-Pictet-Spengler (HIPS) ligation are two examples of such conjugations. Using this technology, related antibodies developed by Redwood Bioscience have entered clinical trials.
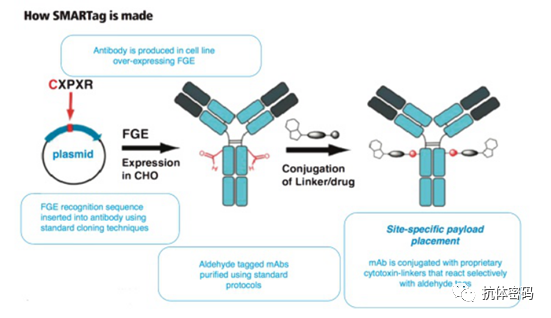
SMARTag™ Platform
2.4 Enzyme-Catalyzed Reactions
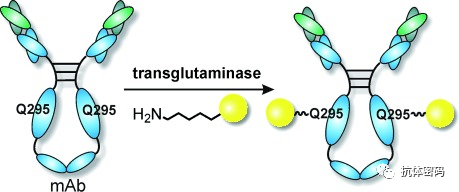
Using enzyme-catalyzed covalent coupling, some researchers have utilized transglutaminase and sortase (sortase) for site-specific conjugation of antibodies. Transglutaminase can catalyze the formation of amide bonds between glutamine and primary amines. The Schibli team was the first to utilize bacterial transglutaminase for site-specific conjugation, but their research found that, despite the presence of abundant glutamine in the antibody, the enzyme-catalyzed reaction only occurred at the Gln295 site in the antibody after the N297 site’s sugar was removed or an N297S mutation was made. Other researchers introduced short peptide tags containing glutamine, LLQGA, which can form ADCs with a DAR of 1.8-1.9 under the catalysis of transglutaminase.
Using a similar approach, sortase (sortase) can also catalyze reactions on specific amino acid sequences, allowing antibodies to be genetically engineered with LPXTG short peptide tags for site-specific conjugation.
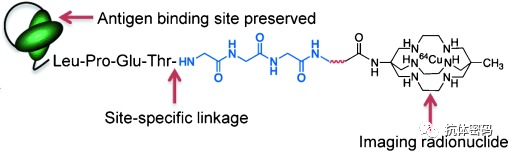
2.5 Introduction of π-Clamps
In addition to using enzyme-catalyzed reactions, some researchers have introduced specific amino acid sequences at the C-terminus of the antibody heavy chain, such as FCPF, which contains a “π-clamp” that provides a unique environment conducive to site-specific modification of cysteine (for peptides containing a “π-clamp” sequence, when part of the “π-clamp” undergoes a gly mutation and reacts under competitive conditions, it will preferentially select those peptides containing the “π-clamp” sequence for quantitative reaction).
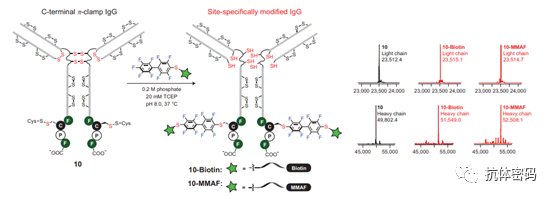
Summary: Overall, second-generation site-specific conjugation technology introduces bioorthogonal molecules into antibodies and utilizes the introduced unique sites for site-specific conjugation. However, the introduction of these unique sites often requires modifications to the antibodies, which may alter some properties of the antibodies and even affect their folding or stability, thus posing challenges for subsequent generation research.
Third Generation Antibody Conjugation Technology
Although second-generation antibody conjugation technology can achieve site-specific conjugation of antibodies, it requires special modifications to the antibodies, which may pose new challenges for the development of ADC drugs. Therefore, some researchers have begun to consider site-specific coupling of natural antibodies without altering their integrity. Third-generation conjugation technology mainly focuses on selecting unique amino acid sites and utilizing unique molecules or proximity effects for site-specific coupling, which can be divided into the following four categories: 1) Interchain disulfide bond modification; 2) Glycosylation modification; 3) Chemoselective modification; 4) Proximity effect-based modification.
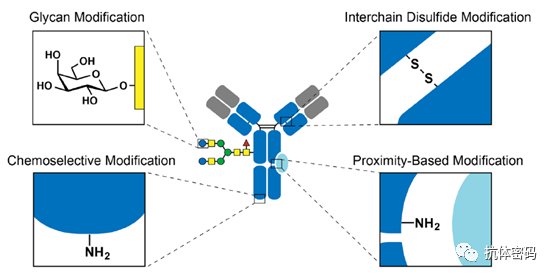
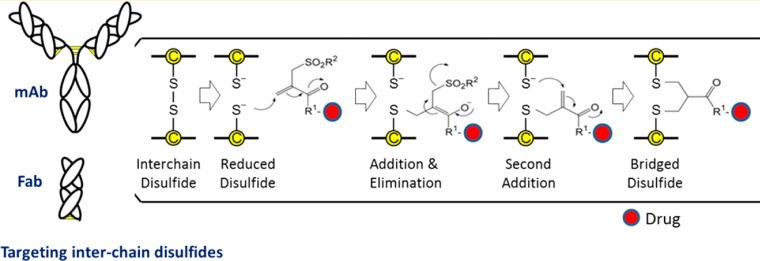
3.1 Interchain Disulfide Bond Site-Specific Conjugation
Utilizing cysteine to couple the payload to antibodies requires a reaction between the reduced thiol in the antibody and the electrophilic species in the payload. Compared to other lysine-based coupling methods, this method has certain advantages, as there are fewer cysteines in antibodies, allowing for higher specificity in conjugation. The four pairs of interchain disulfides can be reduced to eight free thiols, resulting in a uniform ADC with a maximum DAR of 8. The disulfide reconnection technology was developed by the Smith and Lawton research team in 1990, which allows for site-specific coupling without affecting the natural structure of the antibody. It mainly consists of four steps: (1) reducing disulfide bonds to obtain two active cysteines; (2) reacting the reconnection reagent in the payload with one of the cysteines; (3) the remaining cysteine reacts with the cysteine-reconnection reagent complex generated in the previous step to form a new linkage.
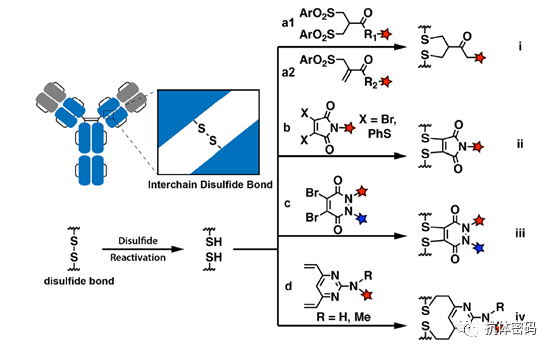
3.1.1 Mono-/Bisulfide Reagents
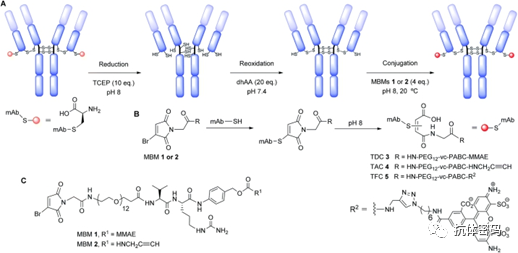
For bisulfide reagents, it first needs to be activated by removing one sulfinic acid salt, and after activation, a monosulfide is obtained that reacts with a thiol in the antibody to form a disulfide bond, while the remaining sulfinic acid salt is removed to obtain a Michael acceptor that reacts with the remaining thiol in the antibody, thus linking the interchain disulfide bonds in the antibody. In 2014, Badescu et al. used this method to couple with trastuzumab antibody, obtaining an ADC drug containing MMAE with a DAR of 4, which also exhibited high stability in a 10 mM DTT reducing environment. (see above image a1) Subsequently, some researchers used the bisulfide reagent alkylating intermediate allyl sulfide for coupling; this reagent has higher solubility, thus exhibiting higher reaction efficiency. (see above image a2)
Monosulfide Coupling
Bisulfide Reagent Alkylation Coupling
3.1.2 3,4-Disubstituted Maleimides
3,4-Disubstituted maleimide coupling is performed using second-generation maleimide reagents (NGMs) such as 3,4-dihalo maleimides and dithiomaleimides. Compared to mono-/bisulfide reagent coupling, the reaction pH is broader (6.2-8), and the reaction rate is fast, effectively reducing the problem of antibody aggregation during the reaction (see below image b). Additionally, its DAR value can be achieved from 0-4 by controlling the dosage and reaction time of the reagents.
3.1.2 Dibromodiphenyl Ketone
Similar to the reaction mechanism of 3,4-disubstituted maleimides, some researchers have utilized dibromodiphenyl ketone for coupling. The two nitrogen atoms in the dibromodiphenyl ketone heterocycle provide possibilities for the coupling reaction, in which the researchers achieved a one-step coupling, thus reducing the impact of related reagents on the antibody during the coupling process (see above image c).
3.1.2 Divinylpyridine
In 2019, Walsh et al. utilized divinylpyridine containing two Michael acceptors to react with reduced disulfide bonds, achieving a high coupling yield. However, because the bridging disulfide bond in this reagent is relatively long (spanning 7 carbon atoms), there is a possibility of disulfide interference during the reaction.
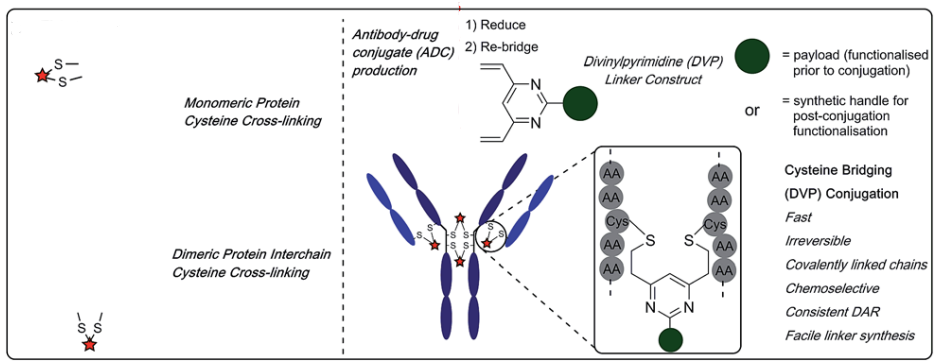
Summary: Currently, many studies have utilized mono-/bisulfides, NGM, and divinylpyridine to achieve specific coupling with the natural antibody’s disulfide bonds, but there are still many limitations and pitfalls. First, disulfide interference during the coupling process is unavoidable, which can affect the efficacy, yield, and scale of production of the products. Although some methods have been developed to improve this issue, such as precise control of reaction conditions, increasing bridging kinetics, and using TECP-linked phenyl thiol derivatives for integrated reactions, these need to be optimized based on the specific circumstances of different drugs. Secondly, under reducing conditions, these bridging reagents cannot distinguish between reduced disulfides and favorable thiols, which limits the lack of free cysteines in the antibody. Wilson et al. reported a trivalent arsenic (As(III)) acid derivative as a potential solution for generating disulfide-thiol orthogonality, as the mono-thiol arsenic acid complex is entropically unfavorable. However, this approach has not been proven in antibody applications, and issues regarding therapeutic-related antibody in vivo toxicity concentrations still need to be resolved. Finally, the poor solubility of these bridging reagents often requires changing the proportions of co-solvents for reactions, increasing the chances of antibody denaturation. If using bisulfide reagents, this issue can be alleviated by using water-soluble monosulfide intermediates, while other reagents can be solved by additionally introducing PEG linkers.
3.2 Glycosylation Conjugation
IgG-type antibodies have two conserved glycosylation sites at N297, located in the CH2 domain, far from the functional domains of the antibody that bind to antigens; thus, utilizing this site for conjugation does not affect the binding function of the antibody. In general, the conjugation using this glycan site primarily involves introducing a new reactive group on the glycan, followed by using related reagents for site-specific conjugation.
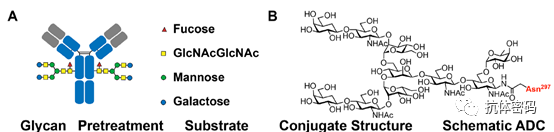
3.2.1 Glycan Oxidation-Based Modifications
Oxidative cleavage of terminal cis-diol groups in oligosaccharides generates an aldehyde group; some reagents containing aminooxy, hydrazine, or hydrazone functional groups can react with it, allowing for the site-specific conjugation of the payload to the antibody. O’Shannessy and colleagues were the first to oxidize the antibody’s glycan using periodate to form aldehyde groups, then react with hydrazone-labeled biotin. Similar methods have been used to couple various molecules, including radioactively labeled organometallic complexes, toxins, and proteins/antibodies. Although these methods successfully achieve site-specific conjugation of native antibodies, the heterogeneity of antibody glycosylation greatly limits the applicability of this method. Additionally, high concentrations of periodate (10-30 mM) can lead to the oxidation of sensitive methionine residues, potentially damaging the antibody’s serum half-life, integrity, and efficacy.
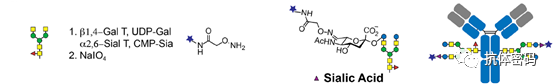
3.2.2 Non-Oxidative Glycan-Based Modifications
To completely avoid periodate treatment, some research groups have performed enzymatic modifications of oligosaccharides by introducing sugar residues with bioorthogonal groups. Bogman et al. demonstrated that using a mutant β1,4-galactosyltransferase GalT (Y289L) can incorporate galactose containing a ketone handle at position C2 into the antibody oligosaccharides. Selective coupling can then occur between the ketone handle and derivatives containing aminooxy functional groups (biotin or fluorescent dyes). However, the conversion rate and efficiency of this process need further improvement. Zeglis et al. applied enzyme-mediated glycan engineering and strain-promoted azide-alkyne cycloaddition reactions to achieve site-specific radioactive labeling of PSMA antibody J591 (see below image). They used β1,4-galactosidase to remove terminal galactose, followed by GalT (Y289L) to introduce azide-modified galactose as the terminal residue of the glycan. They then used chelators containing desferrioxamine (DFO) and employed strain-promoted azide-alkyne cycloaddition reactions to connect to the antibody. Finally, by mixing the chelator-modified antibody with 89Zr, they generated 89Zr-labeled ADCs. The 89Zr-labeled antibodies produced by this method exhibited selective uptake by tumors and demonstrated excellent anti-tumor activity in mouse models of prostate tumors expressing PSMA.
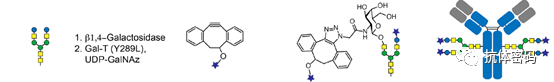
As an alternative strategy, Li and colleagues used Gal T to remodel the antibody glycosylation, transforming existing glycoforms into G2 glycoforms (see image E). They then incorporated sialic acid derivatives containing azide at position C9 into the N-glycans using sialyltransferase ST6GalI. Subsequently, biotin, fluorescent groups, or cytotoxic drugs modified with dibenzocyclooctyne (DIBO) reacted with the azide-containing antibodies, forming antibody conjugates with DAR values of 3.5-4.5. This glycosylation remodeling strategy provides an attractive alternative for preparing homodimeric ADCs with unchanged FcγRIIIa binding. For pharmacokinetic reasons, to achieve a lower DAR, van Delft and colleagues reported a new chemical enzyme coupling strategy to generate ADCs with a DAR of 2 (see image F). After deglycosylation and separately introducing GalNAz through endo-sialidase Endo S2 and galactosyltransferase GalT (Y289L), the modified antibodies can be copper-clicked to effective payloads, resulting in highly stable and homogeneous ADCs with a DAR of approximately 2.
3.2.3 Metabolic Engineering Modifications of Antibody Oligosaccharides
In addition to using enzymatic and chemical strategies for the engineering of antibody oligosaccharides in vitro, it is also possible to introduce sugar analogs containing bioorthogonal reaction groups into ADCs through metabolic engineering of oligosaccharide residues. Okeley and colleagues first reported the use of fucosyltransferase VIII to incorporate fucose analog 6-thiofucose at the terminal N-glycans (see below image). Specifically, 6-thiofucose, screened from about 200 synthetic fucose analogs, can replace fucose in the antibody oligosaccharides with an efficiency of 60-70%. Subsequently, the 6-thiofucose moiety can be coupled with MMAE via maleimide, producing ADCs with an average DAR of 1.3. Compared to ADCs prepared through interchain disulfide/maleimide conjugation chemistry, the ADCs obtained using this strategy maintained improved plasma stability and in vitro anti-tumor activity.

Summary: Despite significant progress in obtaining IgGs with bioorthogonal groups on their glycans, most enzyme-mediated glycosylation engineering techniques remain in the exploratory phase at the laboratory scale. Furthermore, to produce ADCs, some non-natural components must always be introduced into the antibody glycans. Some subtly modified glycans have been reported to have immunogenicity in humans. Therefore, the in vivo stability and immunogenicity of glycan-engineered antibody conjugates still require thorough evaluation.
3.3 Chemoselective Modifications
Conceptually, performing chemical reactions to directly modify specific amino acid residues based on chemoselectivity is the most straightforward method for modifying native antibodies. However, slight differences in the reactivity of different nucleophilic amino acids make it challenging to achieve the desired specificity in chemical modifications. Additionally, due to the heterogeneity of residues between structurally similar antibodies, chemical and regioselective modification methods are typically only effective in specific cases, limiting the generalizability of this approach. A widely used concept is to target lysines with the lowest pKa by precisely controlling the reaction pH; for example, the basicity of the N-terminal amine is weaker than that of other lysines, thus it can be targeted to react under conditions of pH 7.7 ±0.5. Below is a summary of the latest developments in site-specific antibody modifications based on chemoselectivity.
3.3.1 N-Terminal Amine Transamination
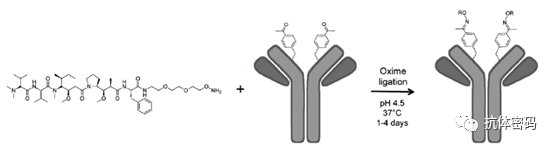
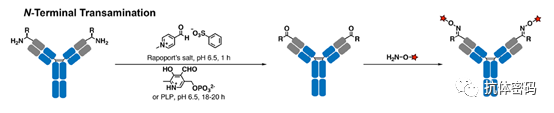
In 2007, Scheck et al. reported the use of pyridoxal-5′-phosphate (PLP, vitamin B6) to convert the N-terminal amine of immunoglobulin light chains into ketones through an amine transfer reaction. The ketones can then be easily functionalized via oxime ligation. Compared to other side chains of lysine residues, the basic N-terminal amine (imine formation) and the more acidic N-terminal α-proton (tautomerization) provide the basis for the N-terminal amine’s chemical selectivity. Moderate conversion rates (47%) were achieved under elevated temperatures (50°C). Based on the same transamination mechanism, Witus et al. selectively converted the N-terminal amine of immunoglobulin heavy chains into ketones using Rapoport salt (RS) under much milder conditions (37°C), achieving an improved yield of 67% (see below image). These amine transfer reagents exhibit some residue preferences; for example, in mouse IgG, PLP prefers light chain aspartic acid over heavy chain glutamine, while RS is more inclined toward heavy chain glutamic acid rather than light chain aspartic acid. Further experiments indicated that the optimal reaction partners for PLP and RS are the tripeptides H-Ala-Lys-Thr and H-Glu-Glu-Ser. Since these N-terminal sequences do not exist in native antibodies, genetic engineering is required to achieve optimal reactivity.
3.3.2 Lysine-Specific Reagents
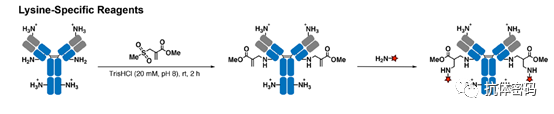
Several attempts have been made to selectively modify certain antibody lysine residues based on controlling reaction kinetics, protein site specificity, or targeting highly reactive lysines. However, due to the unclear mechanism of lysine selectivity, these methods have failed to achieve high yields. Therefore, it has been challenging to apply them to the modification of antibodies or other proteins. To address this issue, Matos et al. reported the use of reagents based on acrylamide sulfonate, combined with pH control, achieving selective and high-yield modifications (>95%) of a single lysine with the lowest pKa (see below image). The sulfonyl oxygen stabilizes the positive charge produced by the amine through a chair-like transition state addition. The smaller polarity of the cysteine S-H group reduces hydrogen bonding interactions, thus explaining the chemical selectivity of lysine over cysteine. The regioselectivity is mainly based on pKa control, as lysines with the lowest pKa are most likely to be reactive under neutral to slightly alkaline conditions. Lysine residues modified with acrylamide can further bind with amine-containing compounds through nitrogen-hetero-Michael addition. The universality of this modification strategy has been successfully demonstrated with five model proteins, including a humanized full-length trastuzumab antibody. Flow cytometry experiments showed that the site-specific modification of trastuzumab with the anticancer drug crizotinib did not affect its binding affinity and specificity towards human HER2. However, if the modified lysine is crucial for antibody function, the conjugation may fail.
3.3.3 Key Point Directed Modifications
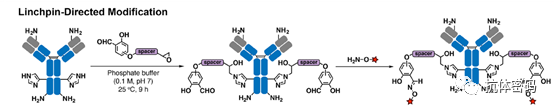
Adusumalli et al. developed another reactivity-based method called key-directed modification (LDM). LDM utilizes fast and reversible amine-reactive groups to guide a slow irreversible reaction between epoxy groups and protein histidine residues (see below image). While a rapid switch equilibrium was established between hydroxymethylbenzaldehyde and lysine residues, the attached epoxide portion would only react with histidine located within a certain distance from the lysine residues. After the irreversible linkage between histidine and the epoxide occurs, the free aldehyde can undergo oxime ligation to conjugate other molecules of interest. Two ADCs, trastuzumab-doxorubicin and trastuzumab-emtansine, have been produced using this technology, exhibiting anti-tumor activity in vitro. To expand the substrate range of LDM, Adusumalli et al. reported another LDM reagent for modifying lysines instead of histidines. By converting the leaving group from the epoxide to 2,6-dibromo-4-(morpholine-4-carbonyl)phenyl ester, more options for substrate optimization are provided.
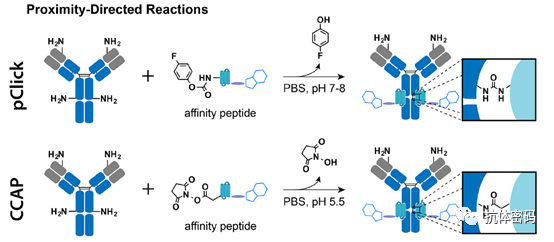
Summary: The selective reactions between probes and specific natural residues (e.g., lysine, cysteine, serine) are becoming an important class of antibody conjugation methods. These strategies enable the preparation of site-specific antibody conjugates without additional antibody engineering or modifications. Currently, efforts based on proximity conjugation are mainly focused on new proximity-inducing chemistries and antibody conjugates that cause minimal disruption to antibody structure and function.
Conclusion
Due to the high therapeutic index and excellent biochemical properties of site-specific ADCs, many efforts are currently focused on site-specific conjugation strategies for antibodies. Numerous methods have been developed for the specific covalent connection of functional molecules to antibodies, most of which require antibody engineering to install unique reactive portions at specific sites, followed by the use of bioorthogonal chemistry to selectively modify these portions. While these technologies provide excellent control over conjugation sites, they have drawbacks such as technical challenges and a lack of generalizability in antibody engineering strategies. Therefore, methods for site-specific conjugation of natural antibodies are more attractive.
Editor: 💧Transparent
This post is intended to convey knowledge. If there are any copyright concerns, please contact us within 30 days of publication.
Original content is prohibited from being reproduced on other platforms without authorization.
If you have any questions, please email [email protected] for more information.
©2021 Medical Overview All rights reserved
