Antibody-drug conjugates (ADCs) are formed by linking monoclonal antibodies (mAbs) with small molecule drugs through linkers. The specificity of mAbs for tumor cells allows small molecule drugs to act effectively within tumor tissues, significantly reducing the high toxicity and side effects of traditional small molecule drugs while improving overall therapeutic efficiency. Currently, there are 13 ADC drugs approved globally, and both domestic and international companies are increasing their investments in this area. According to Grandview data, as ADC drugs continue to be approved and their indications expand to more disease areas, the industry scale of ADC drugs is expected to maintain rapid growth, with the market size projected to reach $21 billion by 2025, and a compound annual growth rate (CAGR) exceeding 50% from 2020 to 2025.

Structure and Mechanism of ADCs
ADCs consist of three components: mAbs that target cancer cells, highly bioactive small molecule drugs, and linkers that connect mAbs and small molecule drugs (Figure 1). After binding to the cancer cell surface antigens, ADCs mediate their entry into cells through endocytosis, are transported via endosomes to lysosomes, where the linker or antibody portion of the ADC degrades and releases the small molecule drug. The small molecule drug then exerts its effect, inducing cytotoxicity and ultimately killing the tumor cells (Figure 2).
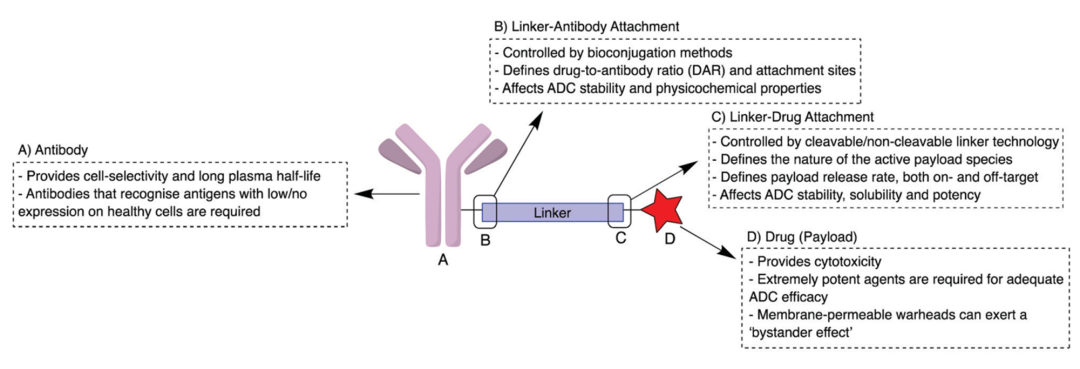 Figure 1 Structure of ADCs
Figure 1 Structure of ADCs
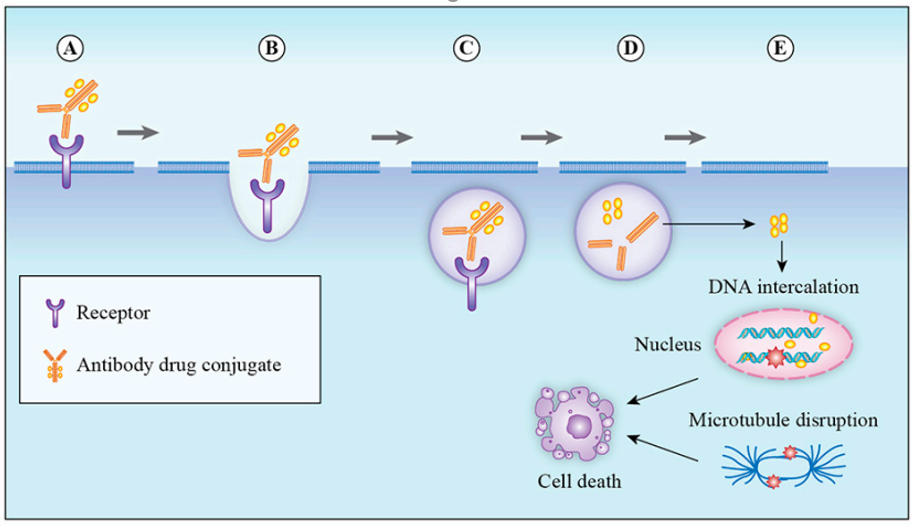
Figure 2 Mechanism of ADCs

Design Requirements for ADCs
In the early 20th century, Nobel laureate Paul Ehrlich first proposed the “magic bullet” theory. Since then, ADCs have made significant progress through years of research. Researchers have found that achieving higher therapeutic efficacy depends critically on the selection of mAbs, small molecule drugs, and linkers within ADCs, and further requirements have been proposed for each of these three components during the design of ADCs.
Antigen Selection:
To achieve lower non-specific toxicity and a better therapeutic index (TI), the antigens targeted by the antibodies in ADCs must be highly expressed in tumor tissues and low or absent in normal tissues. Typically, these antigens are classified into two categories: tumor-specific antigens (TSA) that are expressed only in tumor tissues, and tumor-associated antigens (TAA) that are highly expressed in tumor tissues but low in normal tissues. Furthermore, these antigens need to be expressed on the tumor cell membrane and mediate the entry of ADCs into cells, allowing the small molecule drugs to exert their effects intracellularly. Commonly targeted antigens include CD25, CD33, CD174, etc. (Figure 3).
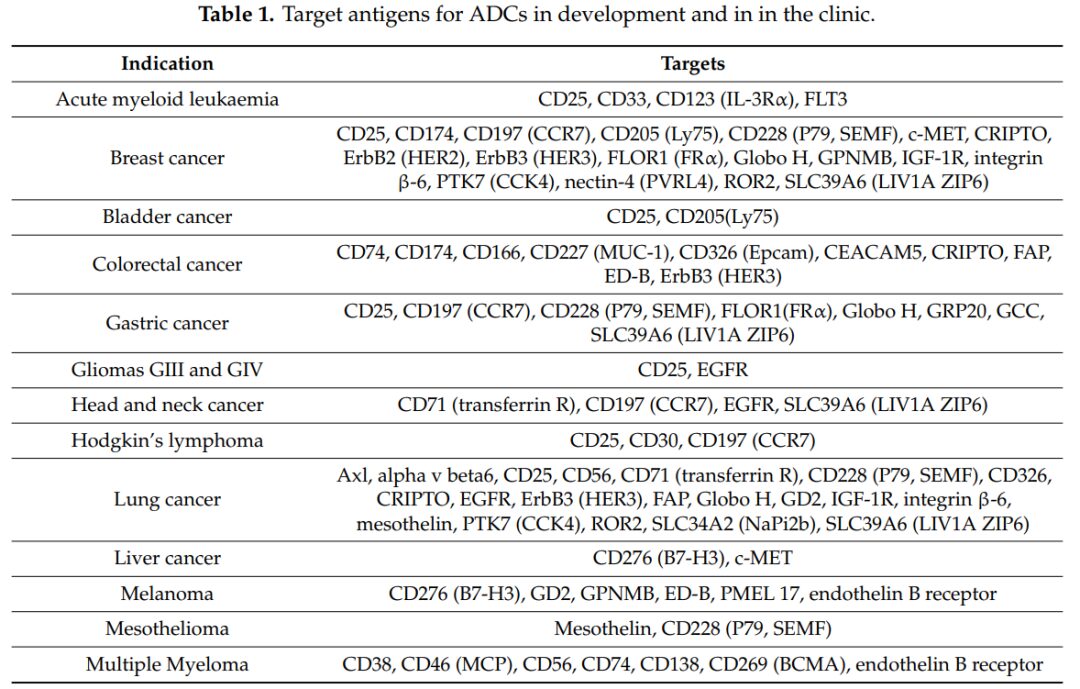
Figure 3 Commonly Used Target Antigens in Research and Clinics
Antibody Selection:
Antibodies must meet requirements for high specificity, strong target binding ability, low immunogenicity, and low cross-reactivity to ensure efficient uptake of ADCs by tumor cells and a longer half-life of ADCs in serum. Currently, ADCs in clinical and preclinical studies often select IgG as the antibody targeting specific antigens. Among them, IgG1 is the most researched and used ADC antibody due to its ability to balance long serum half-life and strong immune activation, along with its high natural abundance. Besides IgG1, IgG4 is also frequently used in ADC designs that require high immunogenicity control due to its lower immune activation effect. Both IgG1 and IgG4 possess 12 intrachain disulfide bonds and 4 interchain disulfide bonds, with the interchain disulfide bonds being highly reactive and commonly used as linker reaction sites. It is worth mentioning that IgG4’s high Fab segment exchange property can lead to off-target effects, so in clinical applications of ADCs using IgG4 antibodies, the S228P mutation strategy is adopted to reduce Fab segment exchange.
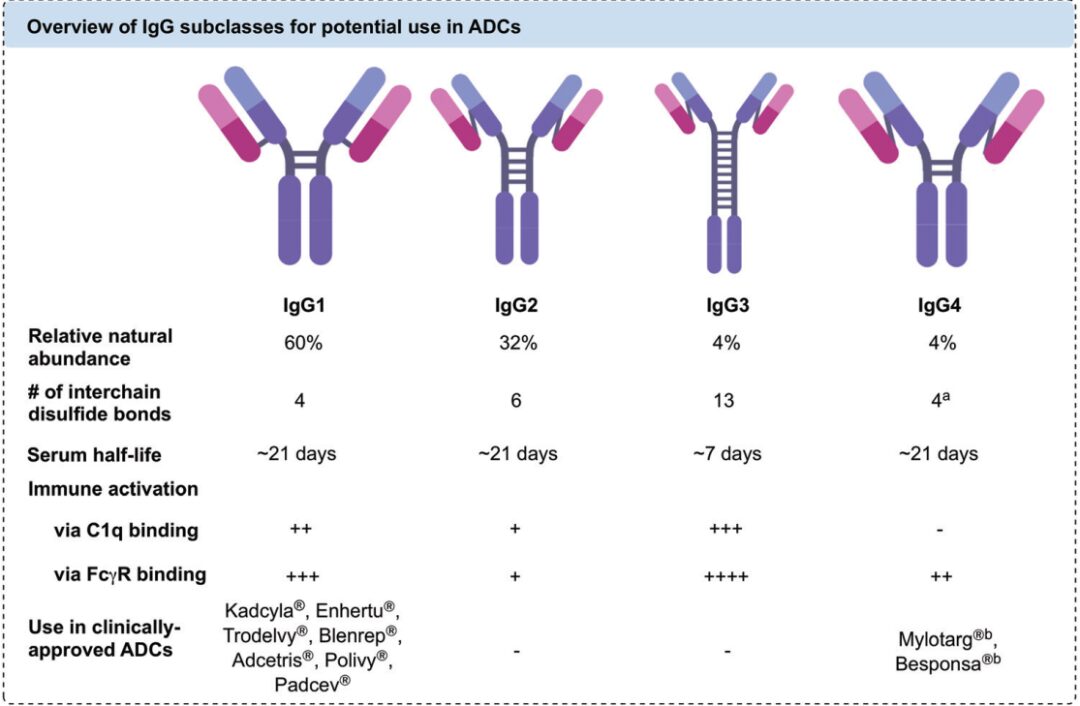
Figure 4 Comparison of Different IgGs
The selection of antibody sources for ADCs has gone through four main stages: murine, chimeric, humanized, and fully human antibodies. Due to issues of high immunogenic rejection, low efficacy, and short circulation half-life associated with murine and chimeric antibodies, humanized and fully human antibodies have become the primary choice in ADC design with the advancement of genetic engineering technology. Currently, the vast majority of clinical and preclinical studies utilize the latter two types of antibodies.
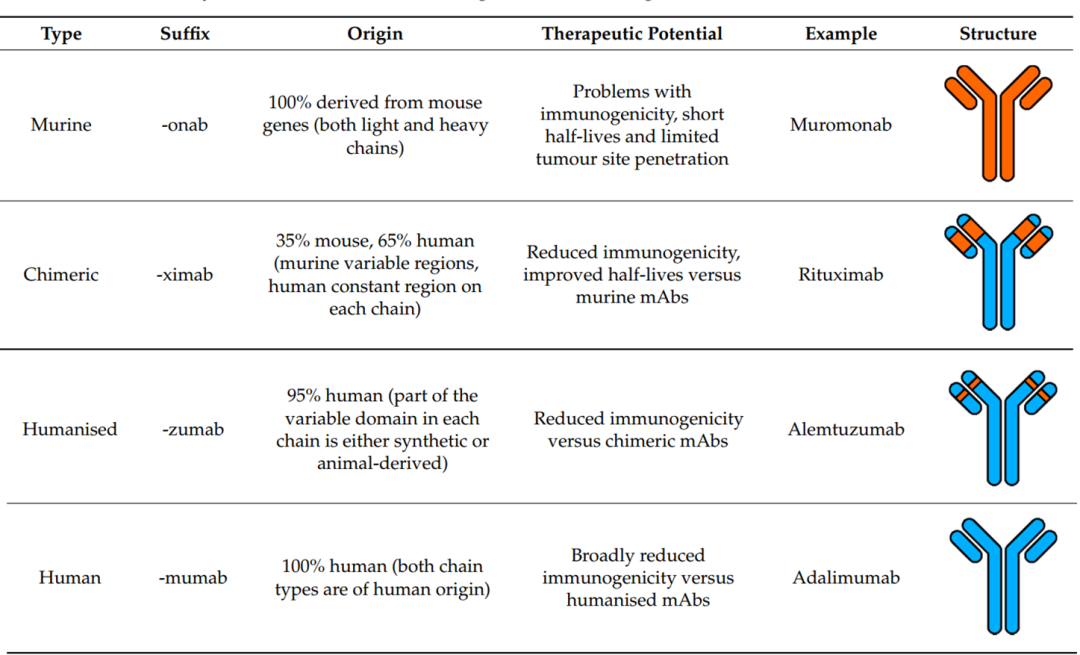
Figure 5 Comparison of Different Antibody Sources (Orange: Murine; Blue: Human)
When selecting antibodies, antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) should also be carefully considered. ADCC and ADCP mediate the immune system’s involvement in the process of killing tumor cells, which can impact the final therapeutic efficacy of the ADC. Therefore, a thorough investigation of the ADCC and ADCP effects of antibodies is essential during ADC design.
Selection of Small Molecule Drugs
Early ADC studies typically selected traditional small molecule chemotherapeutics, including doxorubicin. However, due to the limited number of small molecule drugs that can be conjugated to a single antibody and the limited accumulation of ADCs in tumor tissues, these small molecule drugs with half-maximal inhibitory concentrations (IC50) in the micromolar range do not achieve satisfactory therapeutic effects. Currently, when selecting small molecule drugs, there is often a requirement for IC50 values to be as low as nanomolar or even picomolar levels. These small molecule drugs mainly include two categories: microtubule inhibitors and DNA-targeting chemotherapeutics.
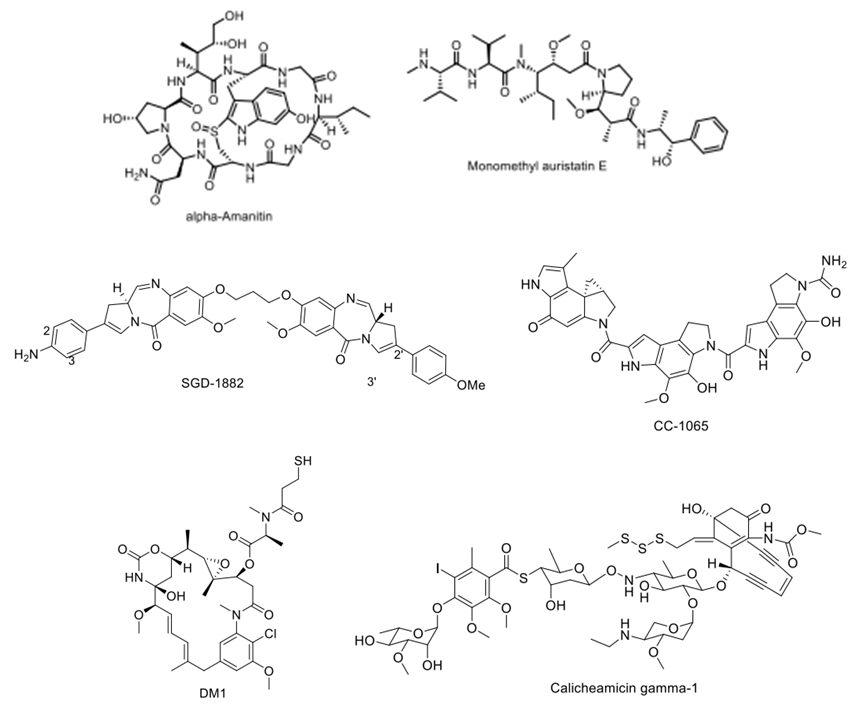
Figure 6 Typical Small Molecule Chemotherapeutics in ADCs
In addition to requiring low IC50 values, small molecule drugs generally need to meet the following criteria: 1. Not easily cause aggregation of ADCs after conjugation to ensure a longer circulation time in the body; 2. Low immunogenicity for both the drug itself and the resulting ADC; 3. Sufficient stability in aqueous solution (blood) and appropriate reactive sites for conjugation with antibodies via linkers, while maintaining biological activity post-conjugation; 4. Can be synthesized through relatively cost-effective processes.
Selection of Linkers
Linkers have a significant impact on the toxicity, stability, specificity, and other properties of the final ADC drug, and the selection of linkers is one of the biggest challenges in ADC development. When selecting linkers, it is essential to ensure that the final ADC can circulate stably in the body for an extended period while releasing small molecule drugs through specific mechanisms after entering cells. Linkers can generally be categorized into non-cleavable types used in first-generation ADCs and cleavable types commonly used in second and third-generation ADCs.
Non-Cleavable Linkers:
Non-cleavable linkers mainly include maleimide-type and thioether-type linkers, which couple small molecule drugs to antibodies by forming amide bonds and thioether bonds. Non-cleavable linkers can ensure high stability of ADCs during circulation in the body. After entering cells via antibody-antigen mediation, they are transported to lysosomes, where the antibody is cleaved by various biological enzymes, releasing the small molecule drug and inducing toxicity. However, ADCs using non-cleavable linkers can generally only target tumors with high antigen expression, as the small molecule drugs released in lysosomes usually carry charged amino acid residues that cannot pass through the cell membrane, thus failing to kill surrounding tumor cells through a bystander effect.
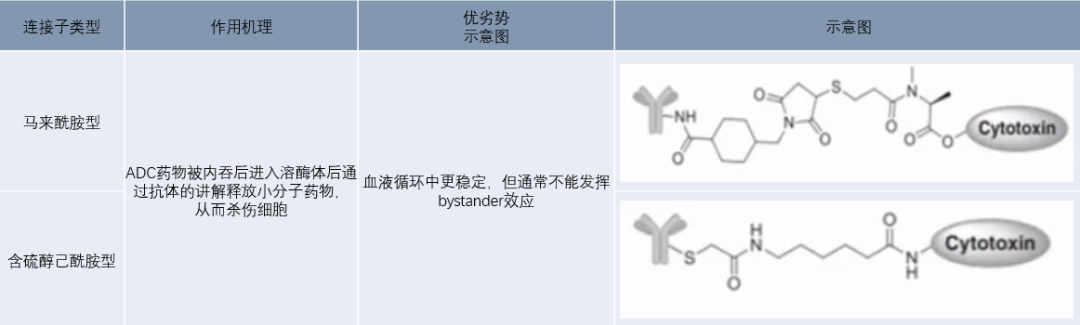
Figure 7 Non-Cleavable Linkers
Cleavable Linkers:
Cleavable linkers are generally divided into pH-sensitive, enzyme-sensitive, and reducible types, which utilize the lower pH of tumor tissues, proteases within tumor cells, and the higher reducing environment within tumor cells to cleave the linkers. ADCs using this type of linker can localize to tumor cells via antibody targeting, accumulate in tumor regions, and release small molecule drugs upon cleavage of the linker stimulated by the unique microenvironment of the tumor, thereby inhibiting cell proliferation and killing cells.
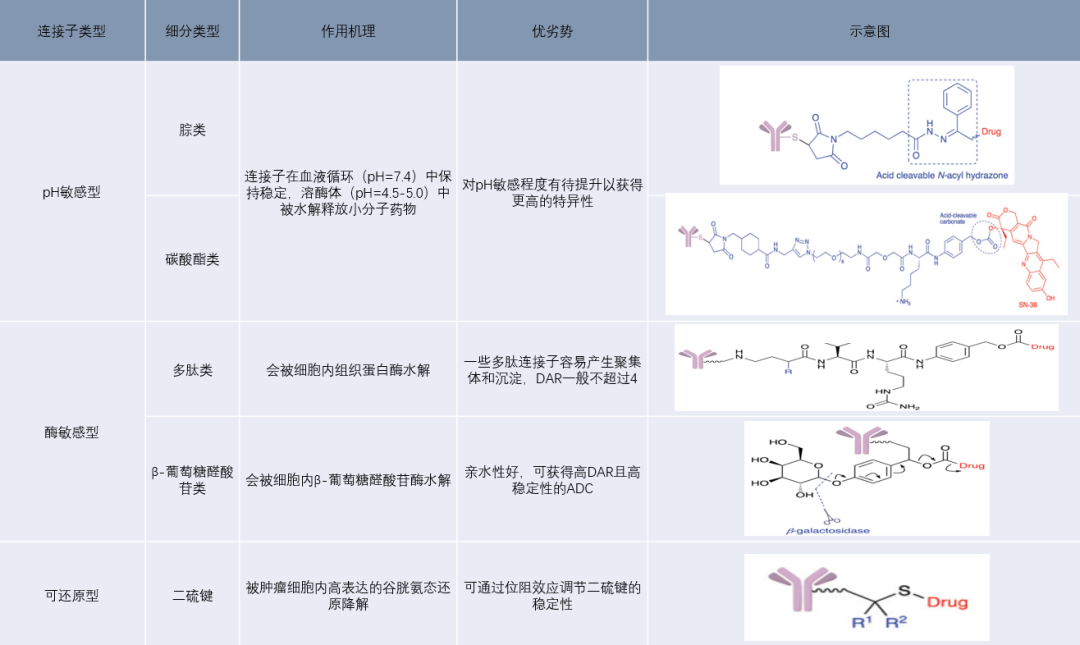
Figure 8 Cleavable Linkers

Drug-Antibody Ratio (DAR)
The number of small molecule drugs linked to a single antibody is defined as the Drug-Antibody Ratio (DAR), which can be obtained through testing methods such as HPLC-MS. A clear DAR value is essential in the later stages of ADC drug development. The limited uptake of ADCs by tumor cells during circulation in the body typically means that a higher DAR is beneficial for increasing efficacy. However, the small molecule drugs used in ADCs often have strong hydrophobicity, and excessively high DAR values can lead to aggregation of ADCs, resulting in reduced circulation half-life and increased toxicity. Therefore, excessively high DAR values are undesirable, with preclinical and clinical ADCs generally having DAR values in the range of 2-8. To achieve higher and more uniform DARs in ADCs, genetic engineering can be used to modify antibodies to have a fixed number of highly reactive sites for conjugating small molecule drugs.
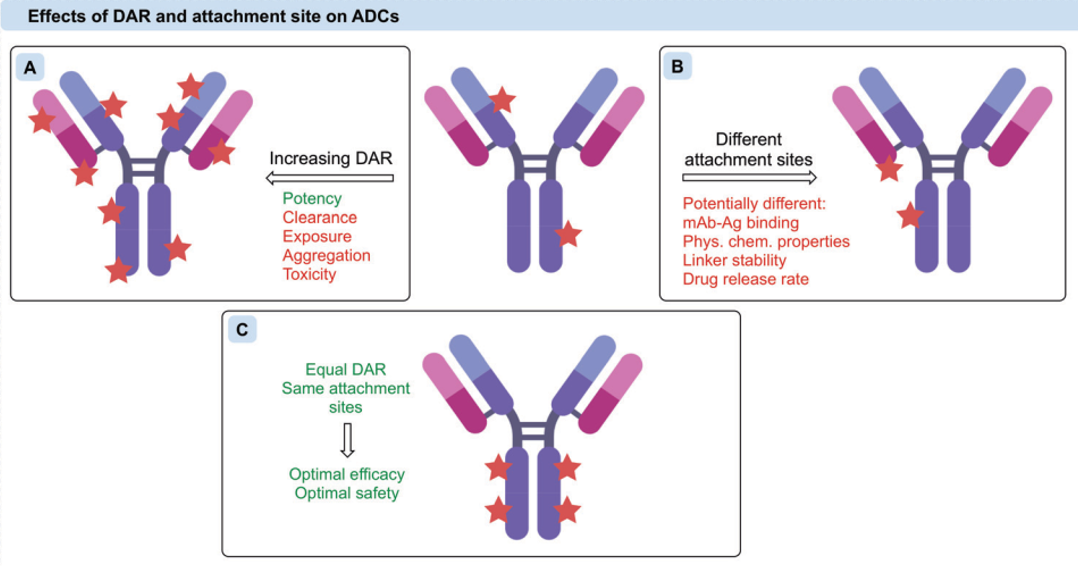
Figure 9 Impact of DAR on ADC Efficacy

Conclusion
ADC drugs combine the advantages of both antibodies and small molecule drugs, providing a more efficient strategy for cancer treatment. Since the introduction of multiple ADC drugs to the market, the response has been positive, and the ADC drug market is vast with great prospects. The ultimate therapeutic effect of ADCs is influenced by multiple factors, including the rational design and selection of antibodies, small molecule drugs, and linkers in the early stages of ADC development, which are the top priorities of the entire development process.
References
Copyright Notice
Personal sharing and forwarding are welcome. Any other media or websites wishing to reproduce or quote the content owned by this website must obtain authorization and indicate prominently “Source: Bai Ao Yun Testing”.
