 Click the blue text to follow us
Click the blue text to follow us
This article is based on the paper “Research on Key GMP Inspection Points in the Production of Antibody-Drug Conjugates” (Author: Ma Yansong, et al.; National Medical Products Administration Center for Food and Drug Review) published in 2025, systematically outlining the entire GMP regulatory chain of ADC drugs from antibody, payload, conjugation, purification to formulation development. It combines the regulations of China, the US, and Europe, practical case studies, and frontline inspection checklists to provide a detailed explanation of each high-risk link and compliance points.
Table of Contents
-
Background of the ADC Industry and the Significance of GMP Inspection
-
Core Links of ADC Structure and Production Process
-
International and Chinese GMP Regulatory Systems
-
Detailed Explanation of GMP Inspection Points in the ADC Process
-
Technical Difficulties and Regulatory Challenges
-
Case Analysis and Common Issues
-
Future Trends and Compliance Recommendations
1. Background of the ADC Industry and the Significance of GMP Inspection
1.1 Industry Status and Market Prospects
-
Global Progress: As of November 2024, 16 ADC drugs have been approved globally, mainly for hematological malignancies and solid tumors, with over 100 products in clinical stages. North America is the largest market, while the Asia-Pacific region (especially China) is growing the fastest.
-
Market Forecast: According to Frost & Sullivan, the global ADC market is expected to exceed $20 billion by 2028, reflecting the significant demand and investment enthusiasm in the field of cancer treatment.
1.2 Production Complexity and Technical Challenges
-
Long Process Chain: ADC involves multiple steps including antibody intermediates, cytotoxic payloads, linkers, conjugation, purification, and formulation, requiring multidisciplinary collaboration at each step.
-
Differentiation Challenges: There is limited experience in conjugation and purification processes, and ensuring batch-to-batch consistency during scale-up is a core challenge across the industry.
-
High-Risk Attributes: There is significant GMP compliance pressure due to highly active raw materials, cross-contamination, operational safety, and environmental protection.
1.3 Strategic Value of GMP Compliance
-
Core Guarantee: A strict GMP system is a prerequisite for ensuring the safety, efficacy, and quality control of ADC drugs, and it is also a threshold for companies entering international markets.
-
Policy Trends: There are significant differences in the implementation levels of GMP in China, and the regulatory side lacks specific regulations, necessitating continuous technological iteration and knowledge updates.
2. Core Links of ADC Structure and Production Process
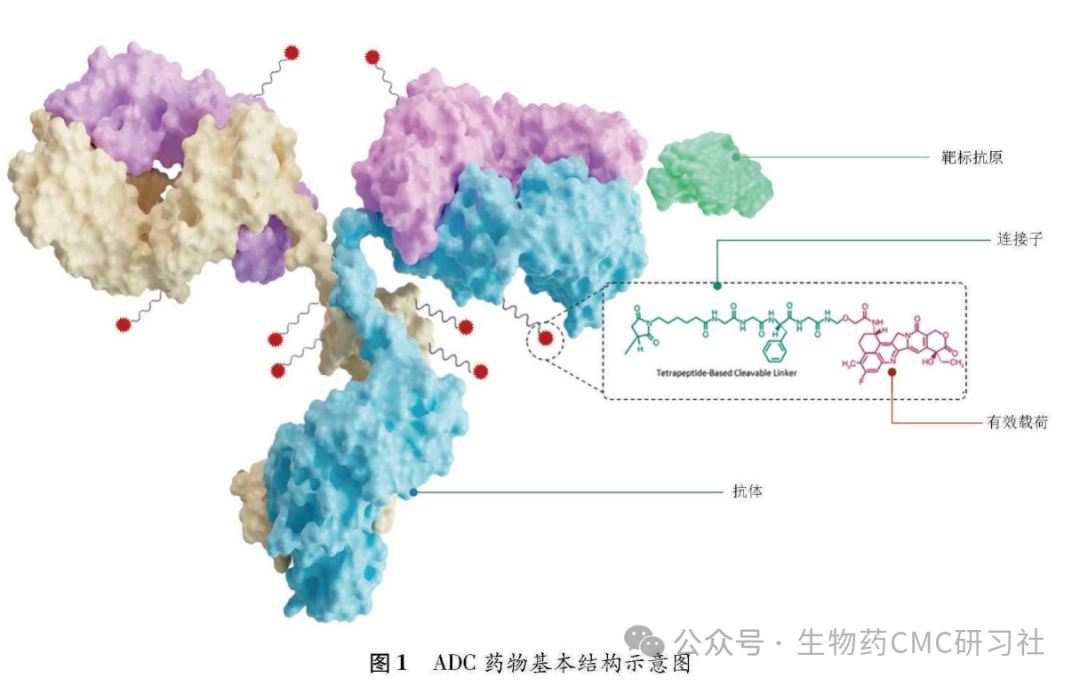
2.1 Molecular Structure of ADC
-
Three Core Components:
-
Antibody (usually IgG): Specifically targets tumor cell surface antigens
-
Payload: Highly efficient small molecule cytotoxins responsible for killing tumors
-
Linker: Covalently connects the antibody and payload, determining the delivery and release mechanism
-
Structural Diagram: Visually demonstrates the ternary relationship between the antibody, linker, and payload, as well as their delivery targets in the body.
2.2 Mechanism of Action of ADC
-
Precise Delivery: Antibody recognizes tumor cells → endocytosis → linker breaks under enzymatic/acidic conditions in the cell → payload is released, killing the tumor while minimizing toxicity to normal cells.
-
Bystander Effect: Released small molecule drugs can also affect surrounding tumor cells that do not express the target antigen, enhancing overall efficacy.
2.3 Comprehensive Process Flow
2.3.1 Production of Antibody Intermediates
2.3.1.1 Upstream Cell Culture
-
Cell Line Construction: Commonly used CHO (Chinese Hamster Ovary) cells, where target antibody genes are introduced through genetic engineering, and high-expression clones are selected. Establish a Master Cell Bank (MCB) and Working Cell Bank (WCB) to ensure traceability, monoclonality, and genetic stability.
-
Cell Expansion and Main Reactor Cultivation: Gradually expand from WCB to the main reactor (e.g., 100L~2000L), monitoring pH, DO, temperature, feeding, stirring rate, etc. Batch-to-batch consistency and abnormal handling are controlled by SOPs.
-
Contamination Control: Aseptic operations are required throughout the process to prevent contamination by microorganisms, viruses, mycoplasma, etc. The components of the culture medium (e.g., animal-derived hydrolysates) must undergo suitability testing, monitoring osmotic pressure, endotoxins, etc.
-
Harvesting and Initial Processing: Cells are harvested when they reach the target density, centrifuged or deep-filtered to collect the supernatant as raw material for downstream purification.
2.3.1.2 Downstream Purification Process
-
Affinity Chromatography (Protein A): Utilizes Protein A to specifically bind to the IgG Fc region, efficiently removing impurities.
-
Virus Inactivation/Removal: Low pH incubation (e.g., pH 3.8, 60 minutes) for virus inactivation, followed by nanofiltration (e.g., 15nm) to further remove viral particles.
-
Ion Exchange Chromatography: Stepwise removal of charge variants, host cell proteins (HCP), DNA, and other impurities to improve purity.
-
Ultrafiltration/Diafiltration (UF/DF): Adjusts the buffer system to provide a suitable environment for the conjugation reaction and concentrates to the target volume.
-
Storage: Antibody intermediates are stored frozen or refrigerated according to project requirements, awaiting downstream conjugation.
-
Critical Quality Attributes (CQA):
-
Purity (>98%)
-
Aggregates (<1%)
-
Protein A Residue (<10ng/mg antibody)
-
HCP Residue (<100ng/mg)
-
DNA Residue (<10pg/mg)
-
Charge variant distribution, glycosylation profile, biological activity (e.g., binding activity, ADCC/CDC)

2.3.2 Synthesis of Payload (Cytotoxin)
-
Chemical Synthesis Route: Multi-step organic synthesis is used based on structural differences, with common payloads such as MMAE, DM1, PBD, etc., involving various organic solvents and intermediates during the reaction process.
-
Purification Methods: After the reaction, high-purity products are obtained through extraction, reduced pressure distillation, silica gel column chromatography, reverse-phase preparative chromatography, and freeze-drying.
-
Impurity Control:
-
Chiral purity (>99%), main peak content (>98%), residual solvents/start materials/by-products must comply with regulatory limits.
-
Special assessment of genotoxic impurities, and Ames or other genotoxicity tests may be conducted if necessary.
-
Personnel and Environmental Protection:
-
Production areas must be independently isolated, operators must wear PPE throughout, and waste liquids and solids must be specially collected to prevent leakage of highly active substances.

2.3.3 Synthesis of Linker-Payload
-
Linking Reaction: Coupling linker (e.g., maleimide, hydrazine) with payload to generate linker-payload intermediates. Reaction conditions must be optimized to prevent side reactions and degradation.
-
Purification Process: Various methods such as chromatography, crystallization, and recrystallization are used for purification to remove unreacted raw materials and by-products.
-
Impurity Classification:
-
Couplable impurities (easily react with antibodies): must be <0.1% to avoid affecting product consistency.
-
Non-couplable impurities (e.g., residual solvents, catalysts): must be effectively removed and proven to meet limits.

2.3.4 ADC Conjugation Reaction
-
Reaction Principle:
-
Specific sites on the antibody (e.g., lysine, cysteine) covalently bind with linker-payload to form ADC.
-
Lysine coupling is mostly random, while cysteine coupling requires reduction of disulfide bonds to expose thiol groups.
-
Key Parameters:
-
Molar ratio of antibody to linker-payload (e.g., 1:2~1:10)
-
Reaction time (1~3 hours), temperature (2~25℃), buffer system (pH 6.0~7.5)
-
Amount of organic solvent (e.g., DMSO) must balance solubility and protein stability
-
DAR Control (Drug-Antibody Ratio):
-
Directly affects drug efficacy and safety. Monitoring through LC-MS, HIC-HPLC, etc., and achieving target DAR (e.g., 3.5~4.0) by adjusting reactant ratios/times/solvents.
-
Case: Excessive DMSO concentration (e.g., >10%) significantly increases DAR, while also raising aggregate levels, thus requiring careful balancing of process parameters.

2.3.5 Purification and Buffer Exchange
-
Removal of Impurities:
-
Using ultrafiltration/dialysis, hydrophobic/affinity/ion exchange chromatography to remove free payload, organic solvents, unreacted antibodies, linker-payload, aggregates, and other impurities.
-
DAR Distribution Optimization:
-
Hydrophobic chromatography can separate different DAR components, collecting the target DAR component to ensure product consistency.
-
Buffer Adjustment:
-
Adjust to the required pH, ionic strength for the final formulation, and add stabilizers (e.g., sucrose, trehalose).

2.3.6 ADC Formulation Development
-
Formulation Design:
-
Referencing antibody pharmacopoeia standards, develop suitable pH, buffer systems, salt concentrations, stabilizers, surfactants (Polysorbate 20/80), considering the hydrophobicity of the payload and the tendency of ADC to aggregate.
-
Lyophilization Process Development:
-
Multi-step lyophilization (freezing, primary drying, secondary drying) parameter development, focusing on the appearance, moisture content, reconstitution time, and solubility of the lyophilized powder.
-
Aseptic Filling and Packaging Materials:
-
Strict aseptic operation procedures (Class A area operations), focusing on the compatibility and leachables risk assessment of packaging materials such as vials, rubber stoppers, and aluminum caps.
-
Stability Studies:
-
Conduct long-term, accelerated, and post-opening stability assessments, focusing on DAR changes, aggregate increases, and the emergence of degradation products.
3. International and Chinese GMP Regulatory Systems
3.1 Core Requirements of GMP System and Its Specificity in ADC Production
3.1.1 Core Requirements of GMP System
-
Controlled Process: GMP emphasizes the systematic, standardized, and traceable nature of the entire drug production process (people, machines, materials, methods, environment, and measurement). The core goal is to ensure drug safety, efficacy, and quality consistency.
-
Deviation Management and Data Integrity: Any deviations, changes, OOS/OOT (out of specification/trend) must have traceability, investigation, and CAPA closure. Data integrity (ALCOA+) is the new norm in international regulation.
-
Process Validation: It is necessary to demonstrate that each process step is not only “effective” but also “repeatable,” as required by continuous process validation (CPV).
3.1.2 Special Applications of GMP for ADC Drugs
-
Multidisciplinary Integration: ADC combines the properties of biological drugs and highly active small molecule drugs, with regulation spanning the fields of biological products, chemical drugs, and sterile drugs.
-
Isolation of High-Activity Areas: The management of highly toxic small molecules such as payloads and linker-payloads differs from traditional antibody workshops, requiring isolated operations, environmental monitoring, and specialized waste disposal.
-
Cleaning and Cross-Contamination Prevention: When ADC production lines share space with other products, specific health risk assessments, residue limits, and cleaning validations are required.

3.2 US FDA GMP Regulatory System
3.2.1 Regulatory Structure and Applicability
-
21 CFR Part 210/211: General GMP requirements for drugs, applicable to all chemical drugs, biological drugs, and ADC products.
-
21 CFR Part 600/610: Specific GMP requirements for biological products, stipulating the production of antibody raw materials, cell bank management, virus safety, etc.
-
21 CFR Part 11: Compliance requirements for electronic records and electronic signatures, applicable to all electronic data systems.
3.2.2 Specific Regulatory Content for ADC
-
Segmented Management of Antibodies and Payloads:
-
Antibody raw materials (including cell banks) are regulated by the Office of Biological Products (OBP).
-
The linker/payload portion and conjugation steps are regulated by the Office of New Drug Quality Assessment (ONDQA).
-
Industry Guidance Documents:
-
“Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use”
-
ICH Q series guidelines (e.g., Q5, Q6, Q7, etc., for biological product quality management)
-
The latest “Clinical Pharmacology Considerations for Antibody-Drug Conjugates” (2024) refines the regulatory aspects of clinical pharmacology and process changes
-
Actual Audit Focus:
-
Process changes require systematic comparability analysis (e.g., cell line changes, conjugation process adjustments), and any changes affecting product attributes must be re-validated.
-
Cleaning validation and cross-contamination assessments must be supported by scientific data.

3.3 European EMA GMP Regulatory System
3.3.1 Regulatory Structure
-
EU GMP Guideline (EudraLex Vol 4): General GMP framework for drug production in Europe, including Part I (human medicines), Part II (active substances), Part III (guidelines and interpretations).
-
Special Technical Guidelines:
-
“Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities”: emphasizes health risk assessments when production lines share space
-
“EU GMP Annex 1” (GMP for sterile products, 2023 version): provides guidance for ADC lyophilization/sterile formulations
-
CHMP’s published specific guidance on ADC pharmacy, clinical aspects, etc.
3.3.2 Regulatory Key Points for ADC
-
Shared Production Risks: ADCs often need to be produced alongside other products, and residue limits must be set based on health risk assessments (HBEL), with strict cleaning validations conducted.
-
Batch Consistency: Detailed comparability analysis with historical batches is required after any changes in each step (e.g., quality attribute drift of the original solution/final product).
-
Stability and Traceability: Batch release and stability monitoring data must be complete and traceable to ensure quality management throughout the product lifecycle.

3.4 China’s NMPA GMP Regulatory System
3.4.1 Basic Regulations
-
“Drug Production Quality Management Standards” (2020 Revision): The fundamental GMP regulation in China emphasizes risk management throughout the process, deviation handling, batch records, etc.
-
“Chinese Pharmacopoeia”: Covers standards for biological drugs, chemical drugs, sterile drugs, etc., serving as the quality baseline for all drugs.
-
Electronic and Data Integrity Regulation: The NMPA has recently released intensive electronic record and data integrity inspection plans, aligning with international standards.
3.4.2 Special Technical Guidance Principles for ADC
-
Core Documents:
-
“Technical Guidance Principles for Clinical Research of Antitumor Antibody-Drug Conjugates” (2023 No. 25)
-
“Technical Guidance Principles for Non-Clinical Research of Antibody-Drug Conjugates” (2023 No. 46)
-
“Technical Guidance Principles for Pharmaceutical Research and Evaluation of Antibody-Drug Conjugates” (2024 No. 14)
-
Regulatory Focus:
-
Standardize the full-chain management of antibody raw materials, payload/linker-payload, conjugation products, and formulations.
-
Emphasize the documentation and scientific nature of the entire process, including raw materials, process changes, analytical methods, impurities, stability, and critical parameters.
3.4.3 Inspection Practices
-
Common Issues in On-Site Inspections:
-
Insufficient evidence for cell bank establishment, missing liquid nitrogen tank alarms/temperature control data
-
Process parameter drift without CAPA root cause investigation
-
Excessive impurities/solvent residues, lacking solvent removal validation
-
Inadequate isolation protection in payload operation areas, high risk of employee exposure
-
Fluctuations in DAR of conjugation reaction batches, lacking consistency analysis
-
Changes in formulation without long-term stability data
-
Recommendations for Enterprises:
-
Continuously compare the requirements of the three major systems: FDA/EMA/NMPA, and continuously optimize GMP documentation and training systems.
-
Establish a self-inspection checklist and dynamic risk assessment mechanism specifically for ADCs.

3.5 Similarities and Differences Among the Three Major Systems and Industry Trends
|
Comparison Dimension |
US FDA |
European EMA |
China NMPA |
|
Regulatory System |
21 CFR series + industry guidelines |
EudraLex + CHMP |
Drug GMP + special guidance principles |
|
Special ADC Guidance |
Yes, continuously updated |
Yes, strengthened in recent years |
Intensive releases in 2023-2024 |
|
High-Activity Area Management |
Emphasizes isolation and validation |
Emphasizes risk assessment |
Emphasizes documentation and validation |
|
Cross-Contamination |
Clear limits/validation |
HBEL + cleaning validation |
Combines risk assessment and on-site verification |
|
Changes and Comparability |
Mandatory systematic validation |
Mandatory systematic validation |
Emphasizes re-validation and batch data |
|
Data Integrity |
21CFR11/ALCOA+ |
EU ALCOA+ |
NMPA special inspections (aligning with international standards) |
4. Detailed Explanation of GMP Inspection Points in the ADC Process
4.1 GMP Inspection of Key Links in Antibody Production
4.1.1 GMP Inspection of Cell Banks
-
Establishment and Traceability of Cell Banks
-
The core goal is to ensure the genetic stability and monoclonal origin of the Master Cell Bank (MCB) and Working Cell Bank (WCB), avoiding cross-contamination.
-
Inspection content includes:evidence of monoclonality, full process records of bank establishment, seed batch traceability, and results of sterility/mycoplasma/virus testing.
-
Storage and Transportation Management
-
Cell banks are generally stored in liquid nitrogen tanks, and during GMP inspections, attention should be paid to temperature records, alarms, and emergency plans for the nitrogen tanks, as well as the sealing of cryovials.
-
Management of off-site storage and integrity of cold chain transportation requires complete validation documentation.
-
Daily Management and Data Integrity
-
Cell inventory operations must be fully recorded, with regular status checks and identity verification to prevent genetic drift due to long-term storage.
4.1.2 GMP Inspection of Upstream Cell Culture Processes
-
Process Parameter Control
-
Inspect production batch process parameters (pH, DO, temperature, feeding, stirring, etc.) records, focusing on abnormal fluctuations (e.g., whether pH curves are consistent, whether cell density and growth curves are stable).
-
Enterprises should have scientific deviation investigation and process change management (e.g., whether root cause analysis and CAPA closure were conducted for abnormal pH or slow cell growth in a certain batch).
-
Culture Medium Management
-
Inspect culture medium batch records, control of animal-derived components, and whether hydrolysates are present; if so, suitability validation data (e.g., stability, osmotic pressure/endotoxin/turbidity of three batches) must be provided.
-
Consumables and Contamination Control
-
Inspect the qualifications, batch traceability, and compliance documents of suppliers for consumables such as cell culture bags, single-use reactors, and tubing to prevent contamination or leachables risks.
-
Verification of Process Testing Results
-
Inspect microbial testing results such as UPB (Unintended Process Bacteria), EOPC (End of Production Cells), protein expression levels, and batch-to-batch fluctuations.
4.1.3 GMP Inspection of Downstream Protein Purification and Quality Control
-
Removal of Impurities and Viruses
-
Inspect the processes and validation reports for affinity chromatography (e.g., Protein A), ion exchange chromatography, and virus removal (e.g., low pH virus inactivation, nanofiltration).
-
Inspect residual impurities (e.g., Protein A, host cell proteins, host DNA, aggregates) testing and limit standards, as well as batch release data for biological load and endotoxins.
-
Consistency of Quality Attributes
-
Inspect charge variant analysis, glycosylation patterns, functional activity (e.g., ADCC, CDC, binding activity) validation methods and batch data.
-
For any attribute drift between batches or after process changes, a comparability analysis report and CAPA management must be available.

4.2 GMP Inspection of Payload and Linker-Payload Synthesis
4.2.1 Raw Material and Supply Chain Management
-
Raw Material Traceability
-
Inspect the qualifications, procurement acceptance records, batch consistency, and compliance documents (e.g., COA, DMF) of key raw materials, reagents, and solvents involved in small molecule synthesis.
-
Supplier Audits
-
Key raw material suppliers should have annual audit reports, and change management processes must be sound; re-validation is required when changing sources or raw materials.
4.2.2 Synthesis Process and Impurity Control
-
Process Validation
-
On-site verification of whether the synthesis process has process confirmation (PPQ), control of key process parameters (e.g., reaction temperature, time, stirring rate), and deviation investigation processes.
-
Impurity Classification Management
-
Couplable impurities (e.g., unreacted linker/payload fragments) must be controlled at very low levels to avoid affecting downstream coupling efficiency and product uniformity.
-
Non-couplable impurities (e.g., solvent residues, catalysts) must be proven to be effectively removed by subsequent purification, with process residue/removal validation data available.
-
Chiral/Genotoxic Impurities
-
Inspect the limit setting and methodological validation of chiral impurities and genotoxic impurities, and provide toxicological safety assessments if necessary.
4.2.3 High-Activity Protection and Operational Safety
-
Personnel and Environmental Protection
-
Inspect the isolation measures in high-activity drug operation areas (e.g., airlock rooms, negative pressure rooms, independent ventilation, HEPA filtration), and whether there are leak monitoring and emergency response plans.
-
Employees must undergo regular health checks, comply with PPE training, and maintain records of exposure incidents.

4.3 GMP Inspection of Conjugation Reactions and Purification
4.3.1 Conjugation Process Parameters and DAR Control
-
Process Validation and Parameter Optimization
-
Inspect conjugation reaction parameters (e.g., antibody concentration, linker-payload ratio, reaction time, temperature, solvent concentration) and target DAR (Drug-Antibody Ratio) control strategies.
-
Reaction KPIs (e.g., conversion rate, aggregate levels, polymer distribution) and endpoint determination data must be available.
-
DAR Distribution Characterization and Analysis
-
On-site inspection of DAR testing reports (e.g., LC-MS, HIC-HPLC), average DAR, and distribution curves, with batch-to-batch data comparisons, and investigations and handling of any exceedances or deviations.
-
Control of Small Molecule Residues and Organic Solvents
-
Inspect whether purification processes such as ultrafiltration and chromatography can efficiently remove organic solvents (e.g., DMSO, acetonitrile) and free payloads, and whether there are release standards for solvent residues in the final product.
-
Inspect experimental data on solvent/toxin removal effectiveness, process robustness reports, and if necessary, conduct solvent compatibility risk assessments.
4.3.2 Purification Processes and Separation Efficiency
-
Target DAR Enrichment and Batch Consistency
-
Inspect whether different DAR components can be separated through graded purification (e.g., hydrophobic chromatography, affinity chromatography), selecting specific DAR components as the final product.
-
Removal of Impurities and Quality Release
-
Inspect impurity removal processes, QC testing data (e.g., aggregates, low molecular weight impurities, unreacted antibodies, linker-payloads), and the release standards and records for the final batch.

4.4 GMP Inspection of Formulation Development and Production
4.4.1 Formulation Development and Process Parameters
-
Formulation Screening
-
Inspect formulation screening records, including buffer pH, ionic strength, stabilizers (sucrose, trehalose, etc.), surfactants (PS-20/PS-80, etc.), anti-adsorption and preservatives.
-
Lyophilization Process Development
-
Inspect experimental development of lyophilization process parameters, including freezing, primary drying, secondary drying temperature/pressure settings, lyophilization curves, moisture content/appearance/reconstitution characteristics, etc.
-
Consistency of Formulation Processes
-
On-site verification of filling, lyophilization/liquid filling processes for cross-contamination and confusion risks, process validation, and continuous monitoring data.
4.4.2 Aseptic and Toxicity Management
-
Aseptic Operation Standards
-
Inspect the cleanliness level of the formulation production area, environmental monitoring, aseptic process simulations (media filling), operating procedures, dynamic monitoring, and deviation investigations.
-
Toxicity Risks and Personnel Protection
-
Inspect protective measures for operations involving payloads, assess cross-contamination risks, protective equipment, and leakage emergency plans.
4.4.3 Packaging Materials and Compatibility
-
Selection of Packaging Materials
-
Inspect compatibility experimental reports of inner packaging materials such as vials, rubber stoppers, and aluminum caps with ADC formulations, leachables, adsorption, and chemical stability assessments.
-
Long-Term Stability Studies
-
Inspect long-term/accelerated/open stability data of formulations, and trends in quality attribute changes of released batches.

4.5 Comprehensive Suggestions and Checklists for Inspection Points (Example)
|
Production Link |
Inspection Items |
Core Focus Points |
Main Documents/Data/Data |
|
Cell Bank |
Establishment/Daily Management |
Monoclonality, sterility/identity verification, temperature control alarms |
Establishment records/validation reports |
|
Upstream Culture |
Process Parameters/Culture Medium |
Process stability, batch consistency, raw material suitability |
Batch curves/culture medium COA |
|
Purification |
Impurity/Virus Removal |
Residual impurities, virus removal, activity testing |
Testing/release reports |
|
Payload Synthesis |
Impurities/Process Validation |
Graded impurity control, process parameters, supplier management |
Validation/batch records/COA |
|
Conjugation Reaction |
DAR/Residues |
Reaction parameters, DAR distribution, aggregates, solvent residues |
Conjugation process/LC-MS data |
|
Purification |
Separation Efficiency/Impurities |
Target DAR separation, impurity removal, release standards |
Analysis/batch reports |
|
Formulation/Lyophilization |
Formulation/Process/Aseptic |
Lyophilization parameters, aseptic filling, packaging material compatibility |
Stability/validation reports |
4.6 Common Inspection Defects and Industry Warnings
-
Insufficient evidence for cell bank establishment, missing liquid nitrogen tank alarm/temperature control data
-
Process parameter drift without CAPA root cause investigation
-
Excessive impurities/solvent residues, lacking solvent removal validation
-
Inadequate isolation protection in payload operation areas, high risk of employee exposure
-
Fluctuations in DAR of conjugation reaction batches, lacking consistency analysis
-
Changes in formulation without long-term stability data
 5. Technical Difficulties and Regulatory Challenges
5. Technical Difficulties and Regulatory Challenges
5.1 Multidisciplinary Integration and Team Capability
-
Core Characteristics: The ADC production process requires full-process collaboration between biotechnology (antibody expression and purification) and chemical synthesis (payload, linker-payload, conjugation reactions), with management models differing from traditional biological or small molecule drugs.
-
Management Challenges:
-
There are significant differences in operational norms, documentation, and risk awareness between the two types of teams, leading to communication gaps.
-
Technical crossover links (e.g., conjugation reactions) require participation from both chemical and biological personnel, leading to unclear responsibilities.
-
Practical Suggestions:
-
Implement interdisciplinary training and establish dedicated ADC positions or multidisciplinary quality review teams.
-
Establish unified GMP standard operating procedures (SOPs) and self-inspection checklists covering regulatory key points from both sides.

5.2 Management of Highly Active Raw Materials and Personnel/Environmental Safety
-
High Activity Risks: Payloads are typically highly toxic small molecules at sub-microgram levels, and linker-payload intermediates also have a high biological hazard level, far exceeding most antibody proteins.
-
Safety Management Requirements:
-
Production areas must have physical isolation, independent airflow, negative pressure ventilation, HEPA exhaust, and online monitoring of air/surface contamination.
-
Employees must wear PPE throughout, undergo regular health checks, and have dynamic control over entry and exit of areas.
-
Leakage incident emergency plans, exposure records, and disposal standards must be in place.
-
Environmental Protection:
-
Waste liquids and waste must be collected and disposed of at high toxicity levels to prevent secondary environmental pollution.
-
Discharge and site testing must comply with regulatory limits.

5.3 Process Complexity and Parameter Control
-
Core Challenges: There are numerous conjugation process parameters (pH, temperature, reaction time, raw material ratios, solvent concentrations, mixing speeds, etc.), with high coupling between each step, and even slight fluctuations in each link may affect the final product quality.
-
Batch-to-Batch Consistency Challenges:
-
Fluctuations in conjugation DAR (Drug-Antibody Ratio) directly affect efficacy and toxicity, requiring precise control.
-
Small changes in raw materials, environment, or equipment status may lead to differences in conjugation efficiency or impurity profiles.
-
Management Measures:
-
Implement QbD (Quality by Design) principles, thoroughly screening critical parameters (CPP) and critical quality attributes (CQA) in advance.
-
Processes for deviation investigation and continuous improvement must clearly define responsibilities.

5.4 Advanced Analysis and Full-Link Quality Characterization
-
Characterization Difficulty: ADC molecules are complex and heterogeneous (different DAR, glycosylation, charge variants), and a single analytical method is often insufficient to cover all CQAs.
-
Analytical Techniques:
-
Joint use of multiple platforms such as HIC-HPLC/SEC-HPLC/LC-MS/MALDI-TOF.
-
It is essential to separately test the quality attributes of antibodies, payloads, linker-payloads, and finished ADCs.
-
Effective payloads, residual solvents, free small molecules, aggregates, etc., must each have separate limits set.
-
Methodological Requirements:
-
All quantitative analytical methods must undergo methodological validation (specificity, sensitivity, accuracy, repeatability, etc.).
-
Detection standards and reference material systems must be sound.

5.5 Diverse Supply Chains and Full-Process Management of Raw Materials
-
Multi-Link Collaboration: Typically, antibodies, payloads, and linker-payloads may come from different production sites or even outsourcing, easily leading to batch inconsistencies, information barriers, and quality traceability issues.
-
GMP Requirements:
-
Strict supplier audits and qualification management, establishing full-chain traceability and batch correspondence.
-
Raw materials entering the factory must have compliance documents such as COA, DMF, and every change must undergo re-risk assessment and validation.
-
Integration Trends:
-
More and more companies are choosing integrated production bases or building their own antibody/small molecule/conjugation/purification/formulation workshops within the same park to reduce supply chain breaks and cross-contamination risks.

5.6 Complexity of Stability Studies
-
Molecular Instability: Payloads are highly hydrophobic, prone to aggregation and degradation, and the stability of conjugated structures is far lower than that of pure antibody products.
-
GMP Management:
-
Comprehensive lifecycle stability studies must be conducted, including finished products (liquid/lyophilized), raw solutions, and key intermediates (e.g., linker-payload).
-
Stability failure modes must be associated with quality attributes (e.g., DAR changes, aggregate increases, emergence of degradation products), and process parameters must be updated in a timely manner.
-
Data Management:
-
Long-term/accelerated/open stability data must be complete and comply with pharmacopoeia and regulatory standards.

5.7 Comparability Studies and Risks of Process Changes
-
Change Challenges: Any change in the ADC process (e.g., cell lines, linker-payload synthesis routes, conjugation parameters) may lead to essential changes in product properties.
-
Comparability Management:
-
Establish comparability assessment systems for “naked antibodies,” “payloads,” “linker-payloads,” “conjugation intermediates,” and “finished ADCs” separately.
-
After process changes, all critical quality attributes must be assessed, and safety/efficacy trials must be re-conducted if necessary.
-
Supply chain changes (e.g., outsourcing to in-house, CMO replacements) also require full-process quality re-validation.

5.8 Material Compatibility and Organic Solvent Adaptation
-
Special Material Considerations:
-
ADC production requires a large amount of organic solvents, and the materials of packaging and equipment must be compatible with organic solvents to prevent leaching, adsorption, and chemical reactions.
-
Single-use reaction bags, ultrafiltration membranes, and tubing that come into direct contact with the finished product must have leachables/compatibility assessment reports.
-
Regulatory Requirements:
-
All materials in direct contact must comply with the limits set by the Chinese Pharmacopoeia, USP, EMA, FDA, and other regulations.
-
Packaging materials must be verified to not release harmful substances during the entire cycle of transportation/storage/use and not affect product stability.

5.9 Industry Practical Suggestions and Future Trends
-
Dynamic Updates to GMP Documentation System: It is recommended that companies update ADC-related SOP/risk control documents at least once a year, continuously absorbing the latest international regulations.
-
Composite Talent Teams: Encourage internal training of multi-skilled personnel in “biological-chemical-analytical-regulatory” fields to promote interdisciplinary integration.
-
Digitalization of Process Platforms: Promote full-process data traceability, automated monitoring, and early warning of risks to enhance batch consistency management capabilities.
-
International Regulatory Coordination: It is recommended that domestic companies actively benchmark FDA/EMA new policies to strive for synchronized registration and inspection recognition internationally.
-
Construction of Process Platforms: Prioritize the establishment of integrated production bases, consolidating the entire process of antibody, payload, conjugation, purification, and formulation to reduce management difficulty.
-
Digitalization of Quality Management: Promote full-process data traceability and automated quality control.
-
Diverse Talent Development: Cultivate teams that understand biology, chemistry, analysis, and regulations.
Conclusion & Copyright Notice
The articles published by this public account aim to promote knowledge and do not constitute investment or patient medication advice. The charts/tables in the text are copyrighted by the original authors.
This article is a first release of original popular science by the “Biopharmaceutical CMC Study Society”.
Industry colleagues are welcome to communicate and correct, and please indicate the source when reprinting.
