
Introduction
Antibody-drug conjugates (ADC) are formed by linking monoclonal antibodies that target specific antigens to small-molecule cytotoxic drugs through linkers, combining the powerful killing effects of traditional small-molecule chemotherapy with the tumor-targeting properties of antibody drugs. Since the first ADC (Gemtuzumab-ozogamicin (brand name: Mylotarg)) was approved for the treatment of CD33-positive acute myeloid leukemia, dozens of ADCs have been developed and approved for cancer treatment.
From selecting the appropriate antibody to the final product, the entire development process of ADCs is a daunting and challenging task. Clinical pharmacology is one of the most important tools in drug development, and utilizing this tool helps to find the optimal dosage of the product, thereby maintaining its safety and efficacy within patient populations. Unlike other small molecules or macromolecules, which usually measure one component and/or metabolite for pharmacokinetic analysis, ADCs require the measurement of multiple components to characterize their PK properties. Therefore, a deep understanding of the clinical pharmacology of ADCs is crucial for selecting safe and effective dosages in patient populations.
Overview of ADC Pharmacokinetics
Pharmacokinetics is an indispensable part of clinical pharmacology and modern drug development. The main purpose of pharmacokinetic studies is to obtain information about drug absorption, volume of distribution, clearance rate, half-life, accumulation after multiple doses, as well as the effects of various disease states, age, weight, and sex on drug pharmacokinetics. These pharmacokinetic parameters can be used to design optimal dosing regimens for patients.
It should be recognized that, unlike small molecules and therapeutic proteins (antibodies or fusion proteins), the PK of ADCs is very complex because ADCs consist of several components. Not only the PK of the monoclonal antibody must be considered, but also the PK of the cytotoxic molecule and the physicochemical properties of the conjugate. Since the molecular weight of the monoclonal antibody accounts for more than 90%, the PK of the different components of the ADC is greatly influenced by its PK. The total antibody (ADC+mAb) PK characteristics provide the best assessment of ADC stability and integrity. The conjugate and the conjugation site also play important roles in maintaining the stability and PK of ADCs. The table below lists the FDA-approved ADCs and their PK characteristics.
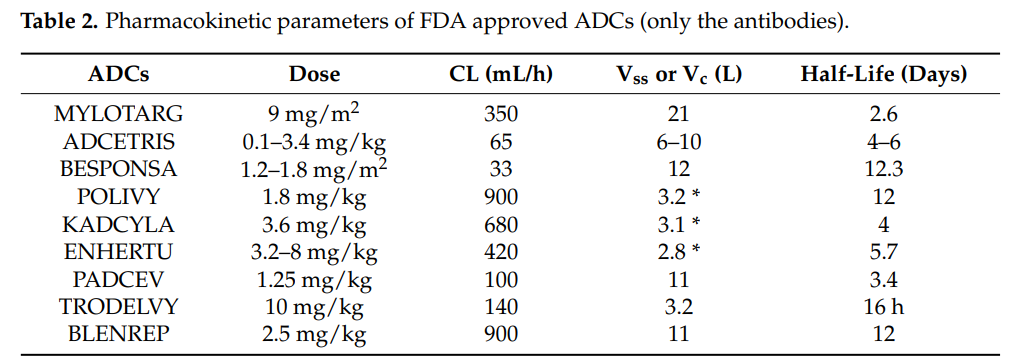
Pharmacokinetic Characteristics of ADCs
Generally, four processes are involved in the body after administration. These processes are absorption, distribution, metabolism, and elimination.
Absorption
Most antibodies are usually administered via intravenous injection or infusion, and antibodies can also be given subcutaneously (SC). However, for ADCs, the current administration route is intravenous injection or infusion. Due to the response to the cytotoxic payload and the local deposition of cytotoxic substances, SC administration may not be suitable for ADCs.
Distribution
The distribution of drugs in the body can be described by the volume of distribution. Due to their size and polarity, the distribution of antibodies and ADCs is usually confined to the vascular and interstitial spaces.
The initial distribution of ADCs is generally limited to the vasculature, and their volume of distribution is typically equal to blood volume. Subsequently, ADCs can distribute into the interstitial spaces. Additionally, ADC distribution can also be affected by target antigen expression and endocytosis.
The distribution and accumulation of ADCs within the same tissue can produce adverse (toxic) pharmacological effects due to the release of cytotoxic drugs or metabolites after ADC uptake.
Metabolism
The degradation/metabolism process of ADCs in the body includes the metabolic processes of the antibody and the small molecule drug. ADCs release effector molecules either within cells (non-cleavable linker) or in the circulation (cleavable linker) before reaching tumor cells, and unbound antibodies and antibody fragments follow the metabolic pathway of antibodies, producing amino acids that are reused by the body.
Free small molecules and/or small molecule drug metabolites linked with amino acid residues may further undergo liver CYP450 enzyme metabolism and may also experience potential drug-drug interactions.
In addition to the properties of the ADC itself, the expression of the antigen, receptor/cell density, FcRn-mediated circulation, Fcγ interactions, receptor-mediated endocytosis, and immunogenicity can all affect the degradation and metabolism of ADCs.
Elimination
ADCs are also eliminated through degradation and excretion. ADCs can enter lysosomes through specific pathways that bind to targets, where they undergo degradation, releasing small molecule drugs before being cleared from the body; they can also be cleared through non-specific pinocytosis, a process involving the neonatal receptor (FcRn) in a recycling process.
ADCs, antibodies, large peptides, and amino acid fragments cannot be excreted through glomerular filtration but are reabsorbed in the form of amino acids. Free small molecules, smaller peptides, and small molecule drugs connected to amino acids can be excreted through glomerular filtration. At the same time, small molecules and metabolites can also be eliminated through enzyme metabolism or excreted into feces via transporters.
Bioanalysis of ADCs
ADCs have several components, and to characterize the PK features of these components, several analytical methods are required, as described below:
-
ELISA immunoassays to determine the kinetics of conjugates and total antibodies;
-
TFC-MS/MS for quantifying free drugs/metabolites;
-
High-resolution mass spectrometry for in vivo drug-antibody ratio (DAR) analysis;
In addition, two types of ELISA immunoassays are used for quantitative measurement of ADC analytes: the first type measures total antibodies, i.e., ADCs with a DAR greater than or equal to zero. The second type measures drug-conjugated antibodies, defined as ADCs with a DAR of greater than or equal to 1.
Other analytical methods include size-exclusion chromatography (SEC) and hydrophobic interaction chromatography (HIC). SEC is the most commonly used liquid chromatography (LC) technique for determining the number of antibody aggregates, and this technique can also be applied to ADCs. Although HIC is a traditional technique for protein separation, purification, and characterization, it is now being used for the characterization and analysis of ADCs.
Cytotoxic Payloads
The cytotoxic payloads of ADCs should possess the following characteristics:
-
The cytotoxic payload should possess appropriate lipophilicity.
-
The target of the payload should be located inside the cell.
-
The payload molecules should be small in size, lack immunogenicity, and be soluble in aqueous buffers to facilitate easy conjugation.
-
The payload should be stable in blood.
Currently, commonly used cytotoxic drug effect molecules include microtubule inhibitors (such as auristatins, maytansinoids), DNA damaging agents (such as calicheamicin, duocarmycins, anthracyclines, pyrrolobenzodiazepine dimers), and DNA transcription inhibitors (Amatoxin and Quinolinealkaloid (SN-38)). Several ADC drugs that have been approved for market use have utilized six different small molecule drugs, three of which use MMAE as the conjugated drug, two use Calicheamicin, and others successfully applied MMAF, DM1, SN-38, and Dxd.
Drug-Antibody Ratio (DAR)
The drug-antibody ratio (DAR) refers to the average number of payload molecules attached to a single monoclonal antibody, typically ranging from 2 to 4 molecules. In rare cases, payloads can safely achieve a DAR of up to 8 using hydrophilic linkers, such as Enhertus and Trodelvys. DAR is crucial for determining the efficacy of ADCs; furthermore, DAR may affect drug stability in circulation, PK, and the toxicity of ADCs.
Studies have shown that ADCs with a high DAR value (7 to 14) clear faster and have reduced in vivo efficacy compared to ADCs with a DAR value < 6. The DAR value and its impact on stability and PK also depend on the conjugation site and the size of the linker.
Lysine or cysteine is typically modified to produce ADCs. Lysine is one of the most commonly used amino acid residues for linking substrates and antibodies, and it is often found on the surface of antibodies, making it easy to conjugate. Mylotargs, Kadcylas, and Besponsas all utilize lysine bioconjugation technology.
Other amino acids such as cysteine and tyrosine can also be modified, with cysteine being modified with maleimide to synthesize ADCs such as Adcetriss, Polivys, Padcevs, Enhertus, Trodelvys, and Blenreps.
Linkers
Linkers are an indispensable part of ADCs, determining the drug release mechanism, PK, therapeutic index, and safety of ADCs. Early ADC linkers were chemically unstable, such as disulfides and hydrazones. These linkers are unstable in circulation, with short half-lives, generally ranging from one to two days. The latest generation of linkers is more stable in circulation, such as peptide and glucuronic acid linkers. The two most common linkers are as follows:
Cleavable Linkers
Cleavable linkers are sensitive to the intracellular environment, releasing free effector molecules and antibodies through degradation and dissociation in the cell, such as acid-cleavable linkers and protease-cleavable linkers. They are usually stable in blood but rapidly cleave in low pH and protease-rich lysosomal environments, releasing effector molecules. Additionally, if the effector molecules can cross membranes, they can eliminate tumors by exerting potential bystander effects.
Non-Cleavable Linkers
Non-cleavable linkers are a new generation of linkers that offer better plasma stability compared to cleavable linkers. Because non-cleavable linkers provide greater stability and tolerability than cleavable linkers, these linkers reduce off-target toxicity and provide a larger therapeutic window.
Immunogenicity
In 11 clinical trials targeting 8 ADCs, the baseline incidence of ADAs ranged from 1.4% to 8.1%, with post-baseline ADA incidence ranging from 0 to 35.8%, figures that fall within the range of therapeutic monoclonal antibodies. Overall, the incidence of ADAs for ADCs is lower in patients targeting hematological malignancies than in those targeting solid tumors; most ADAs are directed against the monoclonal antibody domains of ADCs. Furthermore, in most patients, the semi-antigenic structures of these ADCs do not pose a greater risk of immune response than therapeutic monoclonal antibodies.
Pharmacokinetic Models of ADCs
The application of modeling approaches can integrate PK, efficacy, and safety data to meet the needs of different stages of ADC drug development, such as target selection, antibody affinity, linker stability, animal-to-human extrapolation, dosage selection and adjustment, exposure-response relationships, DDI studies, and so on. Due to the multiple elimination pathways of ADCs (dissociation and degradation) and the complex PK characteristics involving multiple analytes, the kinetic models are also quite complex.
Different models have different applications; for example, a two-compartment model and PBPK model can describe ADC stability characteristics using parameters such as clearance rate, dissociation, and metabolic rates. Currently, non-compartmental models, population pharmacokinetic models, mechanism-based models, and physiology-based models are all applied in ADC pharmacokinetic research.
Conclusion
In the development process of ADC drugs, clinical pharmacology plays a very important role. Through the continuously evolving bioanalytical techniques, comprehensively elucidating the PK/PD characteristics of ADC drugs is crucial for promoting the development of low-toxicity and highly effective ADC drugs. ADC drugs are bound to demonstrate even stronger advantages in the field of cancer treatment.
References:
1. Clinical Pharmacology of Antibody-Drug Conjugates. Antibodies (Basel). 2021 May 21;10(2):20.
Source | Little Medicine Talk
 Pharmaceutical Media Business CooperationMedia PR | News & Conference ReleaseManager Zhang: 18600036371 (same WeChat number)
Pharmaceutical Media Business CooperationMedia PR | News & Conference ReleaseManager Zhang: 18600036371 (same WeChat number)
Click below “Pharmaceutical Media” to follow more exciting content
Disclaimer
“Pharmaceutical Media” public account reprints this article from other public account platforms, mainly to share industry-related knowledge and convey the latest information. The images and articles are copyrighted by the original authors. If there is any infringement, please inform us promptly, and we will delete the relevant information within 24 hours.
WeChat public account push rules have changed again. If you don’t click “Looking” or haven’t set it as a “Star Mark”, we might just disappear into the vast sea of articles~ Click here, don’t miss the latest news from Pharmaceutical Media!👇👇👇