Since the end of 2019 and the beginning of 2020, I have been looking at ADCs, and I have discussed with many experts in the field, leading to many misunderstandings, and my understanding has been continuously refreshed. Many articles about ADCs exist, but many of them contain misunderstandings. Now, there are more successful and failed ADCs and PDCs, and a lot of data is available for us to study and think about. I have always wanted to organize some unwritten understandings. Therefore, this article is particularly long, and I will try to write it in a more accessible way. (Additionally, I do not write for companies, nor do I engage in business collaborations; this account is just for fun.)
Biology is an experimental-driven discipline, so aside from facts, everything else is a hypothesis. It is very important to distinguish between fact and opinion. Therefore, carefully thinking through and interpreting facts is much more important than just listening to opinions. Regarding this field, I will share some of my rudimentary understandings. In the future, some companies in China will definitely be able to do ADCs well. In fact, there are already a few companies that are doing quite well; don’t ask which ones, as I am guessing.
I won’t write about the history of ADCs; it’s an old concept, but until the data from 8201 was released, not many people paid attention to it. I remember calling a leader of a major pharmaceutical company in China at the end of 2019. He thought ADCs could not achieve a large therapeutic window, so they had reserves but had not developed them. Of course, they are now chasing after it frantically.
Will ADCs become like PD1? It is clear that they have already started to resemble PD1. Originally, I didn’t think ADCs would be like PD1 because many times, without a rationale, changing them easily leads to a “me worse” scenario, whether it’s to circumvent patents or to make “me better”.
In the past few years, there weren’t many people with a rationale, but as more trial and error occurred, some understanding has gradually been accumulated, leading to more competition. However, it is regrettable that most drugs may be marginal drugs; it’s a first-place game, and the rest will likely lose money and make noise. I don’t know how so many ADCs will be handled by future medical insurance and centralized procurement; perhaps this is, in a sense, a form of wealth redistribution? Losing investors’ money to subsidize patients, but over time, this game will ruin the entire ecosystem because it creates a negative cycle. The ultimate result may be that no one wants to invest in innovative drugs because they are scared of losses.
For the vast majority of companies working on ADCs, I believe that over 90% of them will only have one path: to find unmet needs globally and go abroad to develop globally competitive drugs. In this regard, avoiding competition with 8201 is essential. Globally, if we consider it as targeted chemotherapy, the wave of ADCs is just beginning. In many disease areas, they can gradually replace chemotherapy, even moving towards neoadjuvant and adjuvant therapy.
How to create globally competitive ADCs is a significant challenge for most companies. This is because overseas, especially in the United States, the depth of preclinical and clinical research is much greater than ours, and the environment allows them enough time to explore new mechanisms and new drugs. We are still better at engineering modifications, but our depth of research into mechanisms and understanding of patients and diseases still lags behind, making it difficult to find a striking variety that aligns well from disease selection to target selection to drug design.
However, this does not mean we have no opportunities. No matter what field ADCs are in, even for miraculous drugs like 8201, there is still room for improvement and progress. From an endgame perspective, one day all drugs will be replaced by new drugs (which is why Buffett buys pharma but not biotech).
What will the next generation of ADCs look like? I believe this is a question that everyone working on ADCs should ponder. Next, I will discuss that there are many properties that can be modified, and improving stability is a good starting point. Of course, this stability refers to stability in plasma, meaning that the toxin should not drop off while free, but should be released in the tumor microenvironment or in lysosomes; otherwise, it will affect the efficacy of the ADC.
The most extreme case is from Trodelvy to Dato to SKB264. Trodelvy’s CL2A is so unstable that the drug rapidly disappears from the bloodstream within four hours, essentially equivalent to a chemotherapy. Dato uses tetrapeptide, which is much more stable, and KTHIOL design carbonate linker from Kelun is even more stable than Dato. The toxins used are all derivatives of Topo1 inhibitors, butDAR is different, and the methods and dosages also vary. However, the efficacy and safety data of Dato and 264 are clearly better than Trodelvy’s. But if we compare 264 and Dato alone, we find that 264 is not necessarily better than Dato (264 ORR is slightly higher, but Dato has some patients who are after Trodelvy), including safety (Gr3 percentages are similar, just with different compositions).
This extreme case illustrates that ADCs with poor stability will definitely not work, but if an ADC with acceptable stability can improve stability, whether it will necessarily be better may require case-by-case analysis (different indications and targets will vary significantly). However, this is not a head-to-head comparison because the linkers are entirely different, the drugs are not quite the same, and there are other chemical properties involved. I am also very curious to know what would happen if we merely changed the conjugation method of 8201 without changing the antibody or Dxd. But it seems that not many people are doing this research, and to do it requires a certain strategy and clinical space to experiment.
To be honest, I still have high hopes for site-specific conjugated ADCs. Site-specific conjugation will certainly have space for specific indications and targets. For example, AMAM cleverly chose PSMA; this target is actually very suitable for glycosylation conjugation. It has sufficient absolute expression and high specificity, making it very suitable to pair with non-membrane-permeable toxins like MMAF, which have ICD. Although there are only three patients’ data, PSA50 is 100%, and the space for castration-resistant prostate cancer in the United States is large enough. Now, Lu177 cannot meet the demand, let alone if it is a drug better than Lu177 (but we need to examine its accessibility, as producing and purifying antibodies with non-natural amino acids has some issues).
Actually, the data for 788 is also acceptable. Except that it cannot be increased beyond 1.5mg, the official reason is that 1.5mg/kg has good enough efficacy. In reality, we can see that aside from some unofficial reasons, the data on breast cancer is good. The clinical trial conducted by Zhejiang University showed that in early 2022 at AACR, under a regimen of 1.5gm/kg 3QW with 69 patients, the ORR was 65% (note that it actually included about 10% of patients who were after 8201; management said a small portion could see PR). The Gr3 AE, according to the company’s disclosed data, is in the low double digits, with each item being single digits. However, breast cancer has already been attacked by 8201, so the company cut off the pipeline for survival until recently when it reintroduced the post-8201 story.
Synaffix sold itself cheaply; from a business model perspective, I think this was expected because it is fundamentally first-generation, with low barriers, expensive prices, and already better solutions available. (I still have high hopes for glycosylation conjugation.) The main problem is that Synaffix’s model and strategy have significant issues. As a first-generation glycosylation conjugation, two enzymes and three steps, the core is those two enzymes and the intermediate process. If you help clients develop processes on-site, clients can learn and potentially not need you anymore, or even find a suitable EndoS client to try to develop themselves. So, the only way is to let clients send antibodies over and then conjugate and send them back in a CRO model, but this model does not make sense without a proven platform. The only way is to either create a pipeline and make drugs yourself, or to find the most suitable disease for glycosylation conjugation, but neither has been achieved.
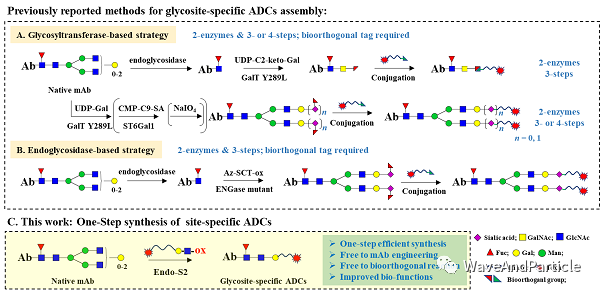
Image from Professor Huang Wei’s paper original link:
https://www.sciencedirect.com/science/article/pii/S2211383521004858
In fact, being able to do glycosylation conjugation with one enzyme in two steps or one-step methods would be better and have lower production costs. Therefore, either like a certain K-starting company that can control glycosylation with N297A, ensuring that the terminal sialic acid is only one type; otherwise, it must be removed for conjugation. Additionally, it must ensure that at least 90% of the sugars are biantennary. If the branching is different, each antibody conjugated will have a different amount of toxin, leading to significant batch-to-batch differences. Or, like Professor Huang Wei has done (I think it’s really impressive), find an EndoS2 that can both cut and transfer, allowing completion in one step, but I don’t know how efficient it is.
In the future, in Her2+ tumors, for a long time, I believe that 8201 will be hard to shake off its leading position. However, we can think about how to improve Her2 low, especially since Her2 low BC represents a large population. Many previously diagnosed Her2 low were classified as Her2 0, yet in this field, 8201 has not reached a level that would discourage exploration. Other areas, aside from Trop2, remain to be explored, such as AMAM’s brilliant choice of PSMA.
————————————————————————————–
What does it take to develop a good ADC?
The concept of ADC is very simple: antibody-drug conjugate. There are essentially three parts, but these three parts are like a lottery because a higher affinity antibody is not necessarily better, nor is a lower one; it’s not always better to have a smaller size (like PDC) or a larger size (like odd dual antibodies). The linker is not necessarily better if it is more stable or less stable, and there are many methods for conjugation, with a greater variety of drug choices, DAR values, mechanisms, toxicities, membrane permeability, ICD, etc. The more terrifying thing is that there is no combination that is a cure-all; different tumors and tissues will have completely different combinations.
This is completely like cooking; adding salt to a cake or sugar to skewers will ruin the taste. But for most people, you don’t even know if you are making a cake or skewers. Some companies attempt to randomly combine elements and solve problems with brute force, which is also very difficult because many things can only be understood in human trials. Therefore, changes should not be made arbitrarily; if changes are to be made, they must be rational. I previously heard someone say to increase the affinity of Trodelvy’s antibody, which was quite frightening. There was also a company that made a slight change to the linker, which caused the drug to be unable to escape the lysosome, ultimately failing in Phase III clinical trials.
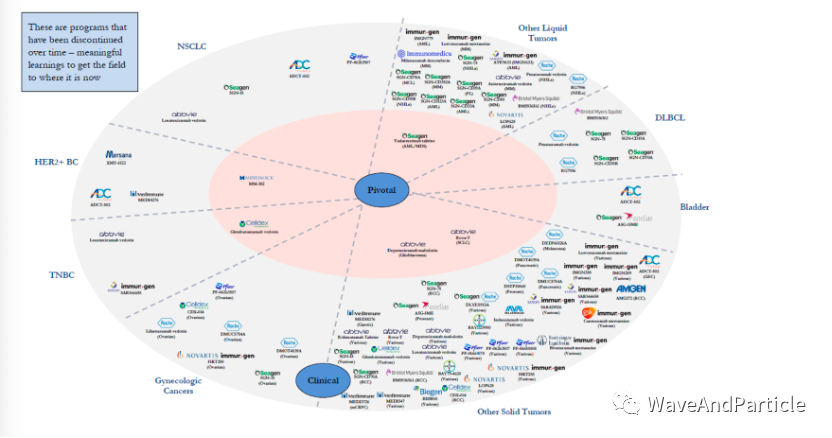
Most people are aware of the successful products, but I will share a few failures for everyone to see how many there are. Of course, most of them failed in Phase I, and very few reached registration clinical trials before failing.
So, what does it take to create a good ADC? Based on my understanding, I will share my insights; please don’t blame me if I am wrong, as I am also guessing.
In general, ADCs are essentially a form of targeted chemotherapy (which may also carry some immune properties depending on the toxin and antibody). Therefore, we hope ADCs can carry toxins into tumor tissues and exert their effects. The more the toxin is exposed in the tumor, the better; the less it is exposed systemically, the better. This increases the therapeutic window for the toxin and allows for the use of some toxins that cannot be directly used in chemotherapy.
In summary, the effective functioning of ADCs requires several steps. After discussing these, we may understand what characteristics a good ADC should embody.
1) After intravenous injection, ADCs must enter our circulatory system, so they undergo a circulation process. We hope ADCs remain stable in the bloodstream, with small molecules and linkers not dropping off, and only releasing in the tumor tissue. Most linker instability is related to the method of conjugation; the earliest lysine conjugation is not to be mentioned, as the later drugs developed by Seagen and others have usedcysteine conjugation. This is better than lysine because an antibody has about 80 lysine residues, and you wouldn’t know where it’s conjugated, as many sites can affect antibody activity. But cysteine conjugation allows us to open disulfide bonds at the conjugatable sites (between light and heavy chains and between the two heavy chains), allowing only eight conjugatable sites.
However, cysteine conjugation also has issues, as the linker is generally a maleimide head, relying on Michael addition. However, in the bloodstream, before the maleimide opens, there will be retro-Michael reactions, causing the entire linker to bind to free thiols in our body, resulting in systemic toxicity. But the most surprising thing is that if we look at FDA reviews, most drugs, including Trodelvy, which drop off upon entry, show very little free payload in the preclinical packages after 96 hours. Why? Because of issues with statistical methodology.
Therefore, most ADC companies, including Seagen, have developed next-generation conjugation methods that are site-specific and quantitative, and more stable in the circulatory system. For example, non-natural amino acid site-specific conjugation (represented by Ambrx and Sutro, with Ambrx being slightly better; Sutro used cell-free synthesized antibodies to circumvent patents, which may lead to issues with post-translational modifications), glycosylation conjugation (represented by Synaffix and Alphamab, with Alphamab being better but harder to achieve—why is it better yet harder? Do your own homework), and enzyme-based conjugation (like Qide and ZW).From the perspectives of production, cost, and quality control, I personally favor glycosylation conjugation, as Seagen’s next-generation ADC also uses glycosylation conjugation.
2) The antibody must be able to bind to antigens on the surface of the tumor.We hope the antibody binds as much as possible to antigens on the tumor surface rather than to those on normal tissue cells. Ideally, the target should be low expressed in normal tissues but highly expressed on tumors, with sufficient absolute expression and a significant difference between the two; there are not many targets that meet this criterion, making Her2 truly the king of targets. There are also some targets that are not perfect but have some of these characteristics, or exhibit such characteristics after certain drug resistances. Here arises a question: what constitutes sufficient absolute expression? I don’t know how much is enough; it seems no one has conducted such research. Different toxins vary in this regard. However, I know that having antigens expressed only by tumors is likely insufficient; currently successful drugs do not have only tumor-expressed antigens, while there are many failures with this characteristic, such as EGFRviii, which AbbVie and another company I forgot attempted and failed, mainly due to efficacy issues. Can we design antibodies that exhibit differential binding in tumors and normal tissues using widely expressed targets? Some mature strategies can be employed, such as designing low-affinity antibodies; some companies have also used other strategies, such as probodies (of course, BioAtla’s AXL Phase II was okay, but Phase III has already failed), but more creative strategies are needed. I won’t elaborate on the creative strategies, as some companies have ideas and data that are quite impressive.
3) Endocytosis.Honestly, I don’t know if ADCs necessarily need to be internalized to exert their effects; some drugs may work without endocytosis in certain tumor types. However, most currently marketed ADCs still rely on endocytosis to function. However, the degree and speed of endocytosis are determined by many proteins, such as caveolin-1. Some ADC resistance has also been attributed to mutations in endocytosis-inducing proteins. Theoretically, we still need to look for targets that favor endocytosis. But this also gives us some inspiration; could certain antibodies (like bispecific antibodies or certain conformational antibodies) be designed to avoid endocytosis in normal tissues but internalize in tumor tissues? A curious thought.
4) Antibody recycling. Since ADCs also contain antibodies, a portion of the antibody will bind to FcRn in early endosomes and may cycle back to the extracellular space before the linker is cleaved, which can affect the release of the toxin within the cell. Therefore, ideally, we do not want ADCs to bind excessively to FcRn. I have seen some papers suggesting that excessive binding to FcRn may also be one of the resistance mechanisms.
5) Linker cleavage and toxin release. After the endosome and lysosome fuse, the linker needs to be cleaved, and the drug must be effectively released. Sometimes we may modify the linker to make it more stable under acidic conditions, requiring a very low pH to effectively release the drug. This suggests that if you don’t understand something, don’t arbitrarily modify it; using mature linkers is a good choice, such as the linker from 8201 or Seagen; new linkers require extensive trial and error.
6) Toxin efficacy. The selection of toxins is very interesting, and I have also thought about it. Different toxins have vastly different characteristics, and many considerations must be taken into account, including mechanisms, resistance, toxicity, and potency in different tissues. Toxins can mainly be classified into three categories: one type is DNA-damaging toxins, the second type is microtubule inhibitors, and the third type includes new toxins (including those affecting transcription, translation, and promoting apoptosis). Cells can die in many ways, and different tissues and cell lines exhibit varying sensitivities to toxins; the impact of toxins on different normal tissues also varies. For example, toxins that affect mitosis will significantly impact rapidly dividing tissues like bone marrow. Typically, the toxicity exhibited by ADCs is related to the toxins rather than the targets.

 From this, we can also infer that most toxicity comes from the systemic release of toxins rather than on-target off-tumor toxicity. After all, truly targeted ADCs are rare. Therefore, returning to the point, addressing the issue of toxins falling off in the circulatory system should resolve most of the toxicity; perhaps target specificity is not as critical as previously thought.
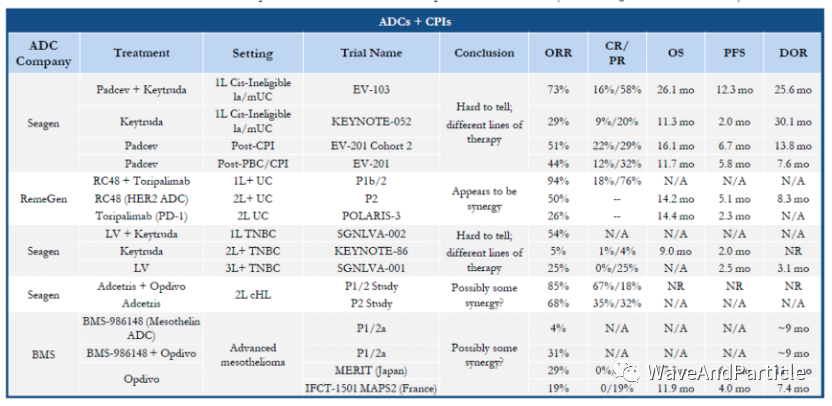
However, when selecting toxins, it is crucial to choose ones that align with the selected tissues and targets because not all tissues will be sensitive to a given toxin. The potency of toxins can vary by dozens of times across different tissues. During the Spring Festival, I read an early paper from Seagen that studied the potency of dxd in different tissue cells. Additionally, if we want to combine with immunotherapy, we need to conduct research on ICD, as not all toxins can induce immunogenic cell death. However, immunogenic cell death can enhance antigen presentation, potentially leading to better synergy with immunotherapy. Below are some clinical trials of ADCs combined with immunotherapy; aside from Rongchang’s, I don’t feel any particular synergy from the others.
From this, we can also infer that most toxicity comes from the systemic release of toxins rather than on-target off-tumor toxicity. After all, truly targeted ADCs are rare. Therefore, returning to the point, addressing the issue of toxins falling off in the circulatory system should resolve most of the toxicity; perhaps target specificity is not as critical as previously thought.
However, when selecting toxins, it is crucial to choose ones that align with the selected tissues and targets because not all tissues will be sensitive to a given toxin. The potency of toxins can vary by dozens of times across different tissues. During the Spring Festival, I read an early paper from Seagen that studied the potency of dxd in different tissue cells. Additionally, if we want to combine with immunotherapy, we need to conduct research on ICD, as not all toxins can induce immunogenic cell death. However, immunogenic cell death can enhance antigen presentation, potentially leading to better synergy with immunotherapy. Below are some clinical trials of ADCs combined with immunotherapy; aside from Rongchang’s, I don’t feel any particular synergy from the others.
 First, a good ADC must have a stable linker in the circulatory system, so I am optimistic about some site-specific conjugations, with a strong preference for glycosylation conjugation. For enhancing the therapeutic window, the selectivity of the antibody may be secondary; the primary focus should still be on the stability of ADCs in systemic circulation.
Second, we need to find a good target. What constitutes a good target? Based on previous conclusions, I believe that sufficient expression in tumor tissues is essential, ideally with a significant expression difference compared to normal tissues. However, if I had to choose between a widely expressed target with sufficient absolute expression and a target expressed only on tumor surfaces, I would prefer the former.
Third, the toxins must be well-researched. Different toxins are suitable for very different scenarios. Many people overlook the study of toxins, thinking that any toxin will suffice; for instance, using DM1 in overseas Phase I trials should yield minimal results. This is because DM1 is a substrate for MDR1, making it particularly prone to resistance, and most fourth or fifth-line patients have likely already developed resistance to DM1. Therefore, the potency, resistance, and mechanisms of different tissues must be thoroughly considered.
Finally, there are some interesting points. Novartis’s PSMA-Lu177 drug is particularly in short supply overseas because Lu-177 has a very short half-life, leading to a limited transport radius, requiring on-demand preparation. Novartis’s drug is actually a PDC; I have always been skeptical of PDCs because they have high Cmax but insufficient AUC, and Bicycle’s clinical results from the previous year were not encouraging. However, I have realized that there are indeed scenarios and diseases suitable for PDCs. For instance, decoupling radiopharmaceuticals is a great choice because radiopharmaceuticals require only a few hours of exposure, and longer exposure is not ideal. In such cases, using PDCs to enhance short-term tumor permeability while reducing systemic exposure is excellent. But what combinations satisfy this condition? Bicycle has also released new data on Nectin-4, which looks promising, along with some detailed data, including the AUC of MMAE in tumors; if interested, you can look it up.
There are also some new directions; with an open imagination, everything can be conjugated.
Pharmaceutical Research Report Sharing Camp
Collecting and sharing mainstream pharmaceutical research reports from the entire market
First, a good ADC must have a stable linker in the circulatory system, so I am optimistic about some site-specific conjugations, with a strong preference for glycosylation conjugation. For enhancing the therapeutic window, the selectivity of the antibody may be secondary; the primary focus should still be on the stability of ADCs in systemic circulation.
Second, we need to find a good target. What constitutes a good target? Based on previous conclusions, I believe that sufficient expression in tumor tissues is essential, ideally with a significant expression difference compared to normal tissues. However, if I had to choose between a widely expressed target with sufficient absolute expression and a target expressed only on tumor surfaces, I would prefer the former.
Third, the toxins must be well-researched. Different toxins are suitable for very different scenarios. Many people overlook the study of toxins, thinking that any toxin will suffice; for instance, using DM1 in overseas Phase I trials should yield minimal results. This is because DM1 is a substrate for MDR1, making it particularly prone to resistance, and most fourth or fifth-line patients have likely already developed resistance to DM1. Therefore, the potency, resistance, and mechanisms of different tissues must be thoroughly considered.
Finally, there are some interesting points. Novartis’s PSMA-Lu177 drug is particularly in short supply overseas because Lu-177 has a very short half-life, leading to a limited transport radius, requiring on-demand preparation. Novartis’s drug is actually a PDC; I have always been skeptical of PDCs because they have high Cmax but insufficient AUC, and Bicycle’s clinical results from the previous year were not encouraging. However, I have realized that there are indeed scenarios and diseases suitable for PDCs. For instance, decoupling radiopharmaceuticals is a great choice because radiopharmaceuticals require only a few hours of exposure, and longer exposure is not ideal. In such cases, using PDCs to enhance short-term tumor permeability while reducing systemic exposure is excellent. But what combinations satisfy this condition? Bicycle has also released new data on Nectin-4, which looks promising, along with some detailed data, including the AUC of MMAE in tumors; if interested, you can look it up.
There are also some new directions; with an open imagination, everything can be conjugated.
Pharmaceutical Research Report Sharing Camp
Collecting and sharing mainstream pharmaceutical research reports from the entire market

Currently: 66 yuan/year, only 0.2 yuan/day
Enjoyover 1400 selectedpharmaceutical research reports
In the coming year, an additional1000 reports will be added