Due to the differences in selecting target antigens, effective payloads, linkers, and conjugation methods for antibody-drug conjugates (ADCs), there is structural diversity and complexity. Moreover, ADCs cannot simply be equated to a combination of antibody drugs and effective payloads, as there are numerous challenges in clinical development, including the complexity of pharmacokinetics, insufficient tumor targeting, inadequate payload release, and resistance.
Today, we will interpret an article related to the clinical research and application of ADCs, aiming to provide a reference for clinical research on ADCs and promote the safe and effective development of ADC drugs.

1. Clinical Development of ADCs
ADCs have pioneered a new paradigm in the development of novel anti-cancer drugs, combining the specificity of large molecule monoclonal antibodies with the potency of small molecule cytotoxic drugs, holding great potential in precision medicine and combination therapy for cancer. Currently, a large number of ADCs are in the oncology clinical development pipeline for the treatment of hematological malignancies and solid tumors. Due to complex pharmacokinetics and pharmacodynamics, the development of ADCs is fraught with challenges. For ADCs to exert therapeutic effects, they must (i) bind to target tumor antigens; (ii) be internalized through receptor-mediated endocytosis; (iii) be transported to lysosomes for drug release; and (iv) diffuse to their targets in the cytoplasm. Any of these steps can lead to failure, and optimizing the following aspects can maximize the likelihood of clinical success: (i) target tumor antigens; (ii) antibody characteristics; (iii) chemical linkers; (iv) cytotoxic drugs; (v) patient populations; and (vi) dosing regimens.
2. Therapeutic Indications
Selecting the ideal therapeutic indication essentially means choosing the best target tumor antigens and patient populations for a specific ADC. Key factors in selecting the best target antigens include specificity, expression levels, internalization capability, target heterogeneity, and accessibility. The specificity of target antigens is a core principle because ADCs are based on targeting tumor-restricted antigens while minimizing delivery to normal tissues. The expression of target antigens in normal tissues relative to tumors further increases the risk-benefit ratio of the drug. The expression level of antigens significantly affects the degree to which ADCs bind to and are internalized by tumor cells. Internalization is crucial for effective cytotoxicity, as ADCs primarily exert their mechanisms of action inside tumor cells. Otherwise, both efficacy and safety may be compromised. Rapid internalization can maintain sufficient expression levels of cell surface antigens, further enhancing the efficiency of ADCs. Antigen expression heterogeneity may exist at the cancer cell level based on tumor type or at the individual patient level. Generally, targets with excessive heterogeneity may limit the extent of patient benefit. Finally, the accessibility of antigens determines how easily ADCs can penetrate tumors. Solid tumors typically present higher barriers than hematological malignancies. The latter are found in blood, bone marrow, and/or lymph nodes, which are areas with higher circulating ADC concentrations. Due to the sparse vascular distribution in tumor necrotic areas, drug penetration in solid tumors may be more challenging.
Choosing tumor types that meet the key factors for target tumor antigens will help prospectively identify the best patient populations. Identifying patients whose tumor target expression levels exceed clinically relevant thresholds (which can be defined in early development studies) will benefit. For certain targets, antibody binding may lead to subsequent depletion of target antigens over time. This primarily occurs in drugs targeting lymphocytes, such as the anti-CD20 antibody rituximab. In such cases, understanding the dynamics of target modulation and re-expression is essential not only for optimizing patient populations but also for optimizing dosing regimens. Many of the most promising ADCs target hematological malignancies, likely due to the presence of cell type-specific targets, the sensitivity of such malignancies to cytotoxic drugs, and the accessibility of most malignant cells to intravenous therapy.
Common target tumor antigens for hematological malignancies include CD33 (acute myeloid leukemia), CD30 (non-Hodgkin lymphoma), CD22 (acute lymphoblastic leukemia, non-Hodgkin lymphoma, B-cell lymphoma), CD19 (non-Hodgkin lymphoma, acute lymphoblastic leukemia, diffuse large B-cell lymphoma), CD74 (multiple myeloma), CD138 (multiple myeloma), CD56 (multiple myeloma, small cell lung cancer, ovarian cancer), and CD70 (non-Hodgkin lymphoma, renal cell carcinoma). For solid tumors, common target tumor antigens include HER2 (breast cancer), GPNMB (breast cancer, melanoma), PSMA (prostate cancer), Lewis y (non-small cell lung cancer), CA6 (malignant tumors), CanAng (solid tumors), Av integrin (solid tumors), SLC44A4 (pancreatic cancer, gastrointestinal cancer), CEACAM5 (colorectal cancer), Nectin-4 (solid tumors), AGS-16 (renal cell carcinoma), anti-Cripto (solid tumors), carbonic anhydrase (solid tumors), mesothelin (solid tumors), TENB2 (prostate cancer), and 5T4 (non-small cell lung cancer).
Typical examples of target tumor antigens that meet key factor criteria are CD30 (non-Hodgkin lymphoma) and HER2 (breast cancer). CD30 is an antigen that is specific, highly expressed, and capable of internalization. It is a member of the tumor necrosis factor family and is not detectable on healthy tissues or resting lymphocytes and monocytes outside the immune system. It is a cell surface marker for Reed-Sternberg cells and serves as a primary diagnostic marker for Hodgkin lymphoma, while also being highly expressed in anaplastic large cell lymphoma and diffuse large B-cell lymphoma. Malignant hematopoietic cells expressing CD30 are sensitive to various cytotoxic drugs. Anti-CD30 monoclonal antibodies can effectively internalize after binding. HER2 is an antigen that is amplified and overexpressed in a small subset of breast cancers. HER2-targeted therapies still have room for improvement due to the presence of acquired resistance. Binding to HER2 receptors triggers HER2 internalization and degradation, supporting the development of ADCs targeting HER2.
3. Transition from Discovery to Early Clinical Development
Advancing ADCs from the discovery phase to early clinical development requires important non-clinical safety assessment projects. Non-clinical toxicology studies supporting new drug applications address numerous issues, such as the selection of animal models, experimental design including dosing and regimens, bioanalytical strategies, and characterization of toxicokinetics and immunogenicity. The safety and exposure data generated from these studies are crucial for advancing ADCs into clinical development. These data are used to determine the maximum recommended starting dose in humans and provide guidance on the exposure range that can be safely evaluated in first-in-human studies or phase 1 trials.
4. Challenges and Considerations in Phase 1 Clinical Study Design
The primary goal of phase 1 clinical studies is to determine a safe and tolerable dose and regimen of ADCs for patients with advanced cancer, which can be used in subsequent phase 2 or phase 3 clinical trials. Secondary objectives typically include preliminary assessments of anti-tumor activity and clinical response, characterization of pharmacokinetics and immunogenicity, and early evaluation of the impact of dose and/or exposure on cardiac QT/QTc interval prolongation. There may also be many exploratory objectives, including assessments of overall survival, characterization of biomarkers, and early evaluation of the relationship between ADC pharmacokinetics and pharmacodynamics.
5. Estimation of First-in-Human Starting Dose
The selection of the phase 1 starting dose for ADCs requires integration of all available non-clinical efficacy, toxicology, pharmacokinetics, and pharmacodynamics data. Since the studies are conducted in tumor patients, the starting dose should be low enough to avoid unacceptable toxicity but not too low, ensuring sufficient systemic exposure that is expected to have pharmacological activity based on non-clinical data. From the toxicology studies supporting ADC INDs, the highest non-severe toxic dose, the dose at which 10% of animals exhibit severe toxicity, or the no-observed-adverse-effect level (NOAEL) is determined. The animal dose is then converted to a human equivalent dose by scaling based on body surface area (as done for small molecule cytotoxic drugs) or by body weight (as done for large molecules such as monoclonal antibodies). If more than one animal species is studied, the human equivalent dose (HED) is derived using the most sensitive species. Finally, a safety factor (i.e., 1/6 of the highest non-severe toxic dose, 1/10 of the standard dose 10, or 1/10 of the NOAEL) is applied to the HED to help protect patients from unexpected toxicity at the starting clinical dose. Given the composition of ADCs, methods for calculating the maximum recommended starting dose (MRSD) have been applied for both large and small molecules.
6. Considerations for Dosing Strategies
Deslandes’ review summarizes clinical pharmacokinetic comparisons of approximately 20 ADCs obtained from phase 1 studies. The dosing methods used are either weight-based or body surface area (BSA)-based. However, fixed-dose administration can also be considered. Weight-based or BSA-based dosing has been used across all tumor drug platforms and is primarily driven by extrapolated doses used in non-clinical animal studies. Weight-based dosing is based on the assumption that the pharmacokinetics of the drug (i.e., clearance and volume of distribution) are proportional to body size. In other words, larger patients require higher therapeutic doses. Additionally, weight-based dosing is believed to reduce inter-patient variability in pharmacokinetics, and controlling pharmacokinetic variability can reduce variability in treatment response. The latter is the most challenging to assess clinically.
For monoclonal antibodies, there is increasing recognition that the relationship between body size and pharmacokinetics is often minimal, and body size can only explain a portion of inter-patient pharmacokinetic variability. Other patient-specific, disease-specific, and physiological characteristics also affect pharmacokinetics and lead to inter-patient variability. The generally accepted approach is that if the predictive impact of body size on pharmacokinetics is greater than 0.5 (representing the exponent in the covariate power model), weight-based dosing will result in smaller inter-patient exposure variability; if the predictive impact of body size on pharmacokinetics is less than 0.5, fixed-dose administration will result in smaller inter-patient exposure variability. If the predictive impact of body size on pharmacokinetics is close to 0.5, both dosing methods are similar in controlling inter-patient variability. These criteria also need to be considered alongside efficacy and safety data.
Pharmacokinetic characteristics of ADCs are determined by their monoclonal antibody components. They have low distribution limited to blood and extravascular spaces, slow clearance, and a long half-life (ranging from days compared to hours for most small molecule drugs). Therefore, it is reasonable to apply the same standards used to evaluate the best dosing for monoclonal antibody therapies to ADCs. To illustrate this, a population pharmacokinetic analysis of ado-trastuzumab emtansine was evaluated. The predictive impact of body weight on the population clearance and central volume of distribution of the conjugated antibody was minimal, at 0.49 and 0.596, respectively. Additionally, the baseline inter-patient variability in clearance and central volume of distribution of the conjugated antibody was low, at 25.6% and 17.5%. Body weight only explained a portion of the variability in clearance, while baseline concentrations of shed HER2 extracellular domain, albumin, trastuzumab, and aspartate aminotransferase, as well as the sum of the baseline values of the longest diameter of target lesions, were also identified as statistically significant covariates. Fixed-dose administration of ADCs has practical advantages over weight-based dosing, including requiring fewer specification configurations, thus reducing drug waste, lowering the risk of dosing calculation errors, and being more convenient in pharmacy or nursing preparation and administration.
7. Optimization of Dosing Regimens
In addition to dosing strategies, the dosing frequency or regimen must also be evaluated. For example, ADCs are administered once every 3 weeks. This approach allows patients to recover from acute off-target toxicities (such as bone marrow suppression) before the next dose. However, more frequent administration of lower doses of ADCs (e.g., once weekly) is also being evaluated. Compared to administration once every 3 weeks, fractionated dosing leads to higher cumulative ADC exposure. This approach may maximize anti-tumor activity while prolonging the occurrence time of acute toxicity and reducing its severity. Fractionated dosing is based on the enhanced concept known as the Norton-Simon hypothesis. In the mid-1970s, a model was derived from clinical and laboratory observations linking the effects of cytotoxic chemotherapy on tumor size to tumor growth kinetics. It was observed that higher doses of cytotoxic chemotherapy caused greater tumor volume regression rates in similar tumor types compared to lower doses. Similarly, more frequent administration of cytotoxic chemotherapy can maintain a certain tumor volume regression rate over a longer period. More frequent dosing may allow tumors to grow less between two treatments and improve eradication effects in a shorter time. Given that ADCs contain cytotoxic drugs, the fractionated dosing strategy can be studied as part of clinical development based on the Norton-Simon hypothesis.
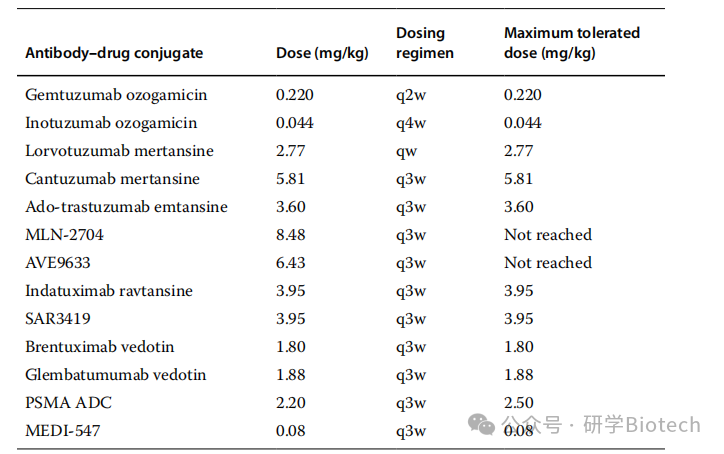
Determining the optimal dosing regimen can maximize efficacy while minimizing toxicity. Table 1 lists the doses, dosing regimens, and maximum tolerated doses (MTD) of 13 ADCs. The most commonly selected dosing regimen is once every 3 weeks at MTD. This decision is based on pharmacokinetic principles, safety and efficacy endpoints, and the convenience of ADC administration in many cases compared to standard care, considering the relatively short elimination half-life of ADCs. Dosing fractionation studies of ADCs based on calicheamicin have shown improved efficacy and reduced toxicity. In addition to PK/PD and target turnover/expression, the combination of efficacy and safety approaches challenges the traditional process of recommending phase 2 doses using classic dose escalation until MTD.
Table 1 Comparison of Doses, Dosing Regimens, and Maximum Tolerated Doses of ADCs
8. Phase 1 Clinical Study Design
Other considerations in phase 1 clinical studies of ADCs include the selection of patient populations and study sites, dose escalation methods, the number of patients at each dose level, the specification of dose-limiting toxicity criteria, the definition of MTD, and the assessments required to determine safety, efficacy, pharmacokinetics, immunogenicity, and biomarkers. Several of these considerations will be discussed in more detail below.
The expression levels of target antigens measured by immunohistochemistry are an important consideration when selecting the best patient populations and tumor types in phase 1 studies of ADCs. In the dose escalation phase, target antigen expression levels are typically not used as inclusion criteria, as the primary goal of this phase is tolerance and safety. At this time, the clinical significance threshold of target antigen expression is usually unclear, and in the absence of data, patients with low or moderate expression levels may still benefit from treatment. During the dose expansion phase, target antigen expression may be used to select participants to assess safety and preliminary anti-tumor activity. Patients defined as having high target antigen expression levels according to IHC standards may respond better to ADC treatment than those with low expression levels. Therefore, the dose expansion phase enriches potential responders, which can guide decisions to directly enter phase 3 trials if early efficacy is confirmed.
Dose escalation is guided by the incidence of dose-limiting toxicities (DLT) as defined by the Common Terminology Criteria for Adverse Events (CTCAE) of the National Cancer Institute. DLT is defined as toxicity that is not attributable to the disease being studied or disease-related processes and may be related to the administration of the investigational drug. The DLT observation period typically corresponds to the length of the first treatment cycle; thus, for ADCs, this period is the first 3 or 4 weeks of administration, depending on the studied regimen. Toxicities occurring during this period are considered acute toxicities, which are essentially hematological or non-hematological. Long-term toxicities, such as peripheral neuropathy or end-organ dysfunction that may occur due to prolonged administration and cumulative exposure to ADCs, cannot be assessed during the standard DLT evaluation period. However, long-term toxicities are included in the overall safety monitoring plan, and their incidence and severity will affect whether ADCs can advance to later stages of clinical development.
Most dose escalation methods in oncology are developed for cytotoxic chemotherapy, with toxicity as the primary endpoint. Given that cytotoxic drugs are a component of ADCs, it is reasonable to apply this dose escalation method in phase 1 studies. Typically, there are two categories of dose escalation: rule-based designs and model-based or adaptive designs. Rule-based designs assign patients to dose levels according to predefined rules based on observed DLTs in clinical studies. These designs do not make any assumptions about the shape of the dose-toxicity relationship but assume that toxicity increases with dose. Examples of rule-based designs include the traditional 3+3 design and its variants, accelerated titration, pharmacologically guided dose escalation, and modified toxicity probability interval methods. Methods can also be combined. For example, dose escalation can start with accelerated titration and then revert to the traditional 3+3 design after meeting predefined stopping rules based on DLT standards. Predefined rules are also used to determine dose levels (e.g., modified Fibonacci sequence) and MTD. Generally, rule-based designs are easy to implement and have successfully identified safe doses for phase 2 clinical studies; however, they do not utilize all available safety information in dose escalation to guide this recommendation. Additionally, compared to adaptive designs, using rule-based designs may treat more patients at doses below MTD.Although adaptive methods are becoming increasingly popular,most phase 1 studies of ADCs use traditional 3+3 dose escalation designs and modified Fibonacci sequences to select dose levels.
For the studies listed in Table 1, dose escalation is typically based on 3+3 or accelerated titration designs. ADCs containing calicheamicin are evaluated at much lower dose ranges, approximately two orders of magnitude lower than those of ADCs containing auristatin or maytansine. ADCs conjugated with DM1 or DM4 show a trend towards higher MTDs. The investigational drugs AVE9633 and MLN-2704 have been reported to be well tolerated, with no MTD reached at the end of dose escalation (>7mg/kg). For other drugs listed in Table 1, a median of six dose escalation levels is required from the first dose level to reach MTD. The most commonly reported dosing regimen is once every 3 weeks; regimens of once every 2 weeks, once every 4 weeks, and once weekly have also been tested. A recent survey of dose levels and efficacy signals in oncology phase 1 trials showed that determining MTD for molecular targeted drugs and conventional cytotoxic drugs requires a median of five dose levels tested, while combination therapies require a median of four dose levels tested. For ADCs, reaching MTD requires a median of six dose levels tested. The need for more dose levels for ADCs may be explained by the selection of starting doses or study design.
Adaptive designs estimate the preset probability of DLT at a dose level by using a model that describes the dose-toxicity relationship, thereby assigning patients to dose levels. These designs use all available toxicity data to ultimately recommend doses and regimens for phase 2 or phase 3 studies. Adaptive designs perform well in estimating the target DLT probability for recommended phase 2 doses without treating too many patients at suboptimal doses; however, they are more challenging than rule-based designs. Modeling and simulation are required, and data from each patient cohort need to be collected rapidly. Additionally, predictions are highly dependent on the sufficiency of prior information used in the model. Examples of adaptive designs include continual reassessment methods and their modifications, time-to-event continual reassessment methods, and Bayesian methods that combine toxicity and efficacy data.
At the end of dose escalation, MTD is selected as the dose that minimizes the difference between the estimated toxicity at the dose used and the preset target toxicity rate (approximately 30%). In the dose expansion portion of phase 1 studies of ADCs (sometimes referred to as phase 2a), more patients are recruited at MTD or a tolerable dose close to MTD to gain more safety experience and collect information on preliminary anti-tumor efficacy, pharmacokinetics, biomarkers, and other endpoints. Data from dose expansion studies can be used to reassess the selected dose or support the selected dose in phase 2 or phase 3 clinical trials.
Phase 2 clinical studies are typically single-arm, open-label trials, as evidence of treatment efficacy can be shown without a control by assessing tumor volume reduction. In a phase 2 study of brentuximab vedotin, the overall response rate in patients with relapsed or refractory Hodgkin lymphoma after autologous stem cell transplantation was 75%, with 34% achieving complete remission. For ado-trastuzumab emtansine, the overall response rate was 25.9% in HER2-positive metastatic breast cancer patients who had previously progressed on HER2-targeted therapy and chemotherapy. Higher response rates were observed in patients diagnosed with HER2-positive tumors. Examples of phase 2 trial designs include the Simon two-stage method, the Bryant and Day method, and the enrichment method. In oncology, the use of randomized phase 2 trials to assess control arms or other dose levels is also gaining attention. Randomized phase 2 trials can better estimate the treatment effect on endpoints for phase 3 trials.
9. Supportive Strategies for Phase 1 Clinical Studies and Beyond
Bioanalytical, biomarker, and clinical pharmacology strategies also support the development of ADCs from phase 1 through to compound registration. Compared to most cytotoxic drugs and monoclonal antibodies, the bioanalytical strategies for ADCs become complex due to their heterogeneous composition and the in vivo changes that occur over time due to decoupling. Multiple detection methods are required, including ligand-binding assays, liquid chromatography-mass spectrometry (LC-MS), and hybrid LC-MS platforms. It is recommended to develop and validate methods targeting small molecule therapeutics and biologics. During the phase 1 stage of clinical development, a comprehensive bioanalytical approach specific to the ADC should be adopted. Various analytes are typically measured, including total antibody, conjugated antibody, antibody-drug conjugate, and unconjugated drug. Each analyte provides unique information about the in vivo behavior of the ADC. Integrating this information is crucial for understanding pharmacokinetics and interpreting the pharmacological effects of ADCs. Subsequently, relationships between analyte exposure and safety and efficacy can be identified during clinical development. Bioanalysis also includes immunogenicity testing of ADCs. In clinical trials, detection of anti-drug antibodies against ADC molecular components is performed using a tiered approach, including detection, confirmation, titer determination, and, where possible, domain-specific characterization. In later stages of development, neutralizing antibody assays may be required to further assess the impact of anti-drug antibodies (ADA).
A comprehensive biomarker strategy begins in phase 1 and continues throughout the clinical development process of ADCs. The goal of this strategy is to find predictive biomarkers that can be used for patient selection and to identify one or more surrogate markers associated with conventional efficacy endpoints (such as best overall response, overall response rate, duration of response, progression-free survival, and overall survival). Many conventional parameters require long-term patient follow-up or are confounded with other factors. ADC-related biomarkers include tumor membrane-bound cell surface antigen expression detected by IHC, shed or soluble cell surface antigens, and circulating soluble markers of anti-tumor activity specific to target patient populations or tumor types. Among these, membrane-bound cell surface antigen expression may prove to be a useful pharmacological diagnostic test. In the phase 2 study of ado-trastuzumab emtansine, patients with high levels of HER2 antigen expression in tumors had better clinical response rates. In phase 1/2a studies, the most common goal is to define relevant clinical thresholds for high membrane-bound cell surface antigen expression and explore the relationship between expression levels and anti-tumor activity.
The biomarker strategy also aims to examine the impact of specific circulating biomarkers on ADC pharmacokinetics. High concentrations of soluble cell surface antigens in the blood may act as a “sink,” binding to ADCs before they reach the tumor. This may lead to target-mediated drug disposition, manifesting as nonlinear pharmacokinetic characteristics, and complicating the selection of appropriate doses for phase 2 or phase 3 clinical trials in phase 1/2a. High concentrations of soluble cell surface antigens in the tumor stroma are believed to act as barriers to efficacy or promote efficacy by improving the tumor microdistribution of cytotoxic compounds, although these concentrations are difficult to measure. For trastuzumab, a naked monoclonal antibody targeting HER2, a minimal physiologically-based pharmacokinetic model was used to describe target-mediated drug disposition (TMDD), considering membrane-bound and soluble HER2 in plasma and stroma. By using this extended TMDD model for simulations, researchers predicted that the trough concentration of trastuzumab was negatively correlated with soluble HER2 serum concentrations, particularly when soluble concentrations were in the range of 500–1000 ng/mL. This corresponds to the clinical HER2 serum concentration range, within which changes in trastuzumab half-life were observed in a phase 2 study. Similar model-based approaches can be used to assess the impact of soluble cell surface antigens on ADC pharmacokinetics and guide dose selection during clinical development.
10. Clinical Pharmacology Considerations
Clinical development paradigms typically characterize the clinical pharmacology of ADCs through the following evaluations of monoclonal antibodies and/or small molecule cytotoxic drugs: (i) phase 1/2a safety/tolerability and pharmacokinetics/pharmacodynamics assessments (as described above); (ii) organ function impairment studies; (iii) drug-drug interaction assessments; (iv) immunogenicity assessments; (v) QT/QTc interval prolongation measurements; and (vi) pharmacology. As shown in Figure 1, these assessments span the entire ADC clinical development process and are guided by data obtained from phases 1 to 3 clinical trials.
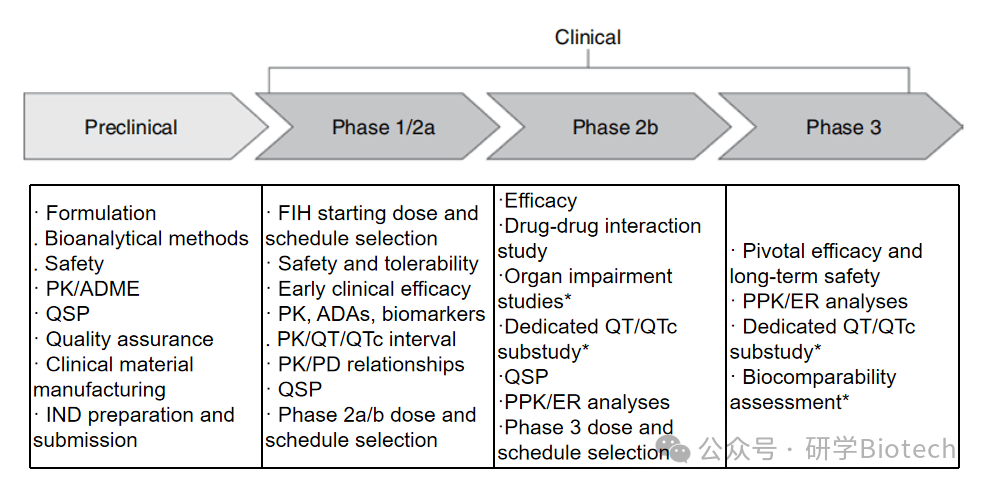
Figure 1 Clinical Pharmacology and Pharmacokinetic Development Strategies for ADCs
11. Assessment of Organ Function Impairment
The necessity of conducting renal function impairment assessments is primarily driven by the extent to which cytotoxic drugs are cleared by the kidneys. Large molecules do not undergo glomerular filtration; therefore, monoclonal antibody-based therapies typically do not require dedicated renal function impairment assessments. The necessity of conducting hepatic function impairment assessments is primarily driven by the extent to which cytotoxic drugs are cleared by the liver. Hepatic function impairment is unlikely to affect the exposure of monoclonal antibodies, as less than 20% of the dose is metabolized in the liver. Therefore, monoclonal antibody-based therapies typically do not require dedicated hepatic function impairment assessments. However, for ADC drugs, if dedicated organ function impairment studies are not conducted, the impact of renal and hepatic function impairment on ADC pharmacokinetics is typically assessed in population pharmacokinetic analyses.
12. Drug-Drug Interaction Assessments
The necessity of conducting drug-drug interaction (DDI) assessments is primarily driven by the potential for cytotoxic drugs to interact with other drugs. However, monoclonal antibodies may also require exploration of DDIs to some extent due to their pharmacological characteristics. The most commonly recorded DDI mechanism for monoclonal antibodies is downstream changes in the expression levels of cytochrome P450 drug-metabolizing enzymes due to cytokine-mediated changes. Interactions typically occur between monoclonal antibodies (the perpetrator) and small molecule cytotoxic drugs (the victim), but due to their differing clearance mechanisms, these interactions are usually mild and uncommon. However, given the narrow therapeutic window of ADCs, thorough DDI risk assessments are crucial for determining the risk-benefit ratio for patients. Early understanding of the degradation and elimination pathways of ADCs, as well as evaluating the cytochrome P450 response phenotypic analysis of cytotoxic drug metabolites, cytochrome P450 inhibition and induction potential, and transporter interactions and inhibition potential through in vitro and preclinical studies, is essential. Typically, this information is combined with the clinical pharmacokinetics and pharmacodynamics of ADCs and their analytes for DDI risk assessments and to determine whether dedicated clinical DDI studies are needed.
13. Immunogenicity
Immunogenicity may occur against any component of ADCs: monoclonal antibodies, chemical linkers, and/or cytotoxic drugs. The presence of anti-drug antibodies (ADA) may lead to changes in pharmacokinetics, safety, and efficacy, which must be assessed throughout the preclinical and clinical development processes. In phases 1 to 3 clinical trials, paired pharmacokinetic and ADA samples are collected at various predefined time points to characterize the baseline, early occurrence, and duration of antibody formation. Pre-dosing samples are preferred, as ADC concentrations are lowest at this time, minimizing the risk of assay interference. To capture the dynamic characteristics of antibody formation, it is recommended to collect samples approximately every 16 weeks, as the half-life of observed endogenous immunoglobulin G antibodies is approximately 21–25 days. The incidence of ADA can provide important indicators for clinical interpretation. Additionally, ADA status can be used as a time-varying covariate in population pharmacokinetics and exposure-response analyses to identify the impact of immunogenicity on pharmacokinetic parameter estimates and efficacy or safety outcomes. The incidence of ADA ranges from 5.3% for ado-trastuzumab emtansine, which contains fully human antibodies, to 37% for brentuximab vedotin, which contains chimeric antibodies.
14. QT/QTc Assessments
The clinical risk of QT/QTc interval prolongation caused by the cytotoxic components of ADCs is very important. The traditional method for assessing cardiac repolarization delay is through thorough QT/QTc studies. Thorough QT/QTc studies are randomized, blinded studies typically conducted in healthy volunteers, comparing the potential for QT/QTc interval prolongation of investigational treatments given at supratherapeutic doses with positive placebo controls. Given their composition, ADCs cannot be administered at such high doses to healthy volunteers, and randomly assigning target patient populations to positive placebo controls is unethical. For these reasons, alternative methods to thorough QT/QTc studies are typically used. One approach is to incorporate robust electrocardiogram and pharmacokinetic assessments into phase 1/2a studies to explore the dose-response and exposure-response relationships of QT/QTc interval prolongation at doses and exposure levels above those expected to be achieved therapeutically. These data can be used to early exclude the potential for QT/QTc interval prolongation and avoid the need for more formal QT/QTc assessments in future clinical studies. If signals of QT/QTc interval prolongation are detected during early development, dedicated QT/QTc studies or QT/QTc sub-studies will be conducted.
15. Pharmacology Strategies
Pharmacology strategies are integrated at all stages of ADC development. Due to the complexity of ADC pharmacokinetics and pharmacodynamics, pharmacological approaches can provide insights not only into ADC disposition and pharmacology but also into associations with biomarker, efficacy, and safety data. As mentioned earlier, several ADC analytes (e.g., total antibody, conjugated antibody, conjugated drug, and unconjugated drug) are measured throughout the clinical development process, and the factors driving efficacy and toxicity may differ, with each analyte contributing differently to physiological and pharmacological effects. Mathematical models can be used to integrate the dynamic relationships between these different ADC analytes and provide opportunities to delineate each analyte’s contribution to efficacy and safety.
The range of pharmacological modeling approaches can vary from relatively simple empirical models to more complex mechanism-based mathematical models that can be used to understand and describe the characteristics of ADCs. Typically, empirical nonlinear mixed-effects models are used to describe ADC disposition and pharmacokinetics through population pharmacokinetics, where demographic and pathophysiological covariates are identified for their impact on pharmacokinetics. In this case, the selection of analytes is crucial. For ado-trastuzumab emtansine, it is believed that the concentration of conjugated trastuzumab drives the efficacy and safety of the compound and is used for population pharmacokinetic analysis based on data generated during clinical development. Bender et al. developed a semi-mechanistic model to further describe the disposition curve of conjugated trastuzumab and the individual drug-antibody ratio (ranging from 0 to 7). Similarly, Gibiansky and Gibiansky developed a TMDD model using clinical data to describe the disposition of ADCs and applied several approximations, including rapid binding, quasi-steady state, and Michaelis-Menten approximations, to derive a simplified TMDD model for ADCs with load-independent characteristics. They then explored the potential for further simplification of the system using certain assumptions and compared the findings of the model with data simulated from the clinical population pharmacokinetic model of conjugated trastuzumab. Lu et al. developed a semi-mechanistic integrated model that connected the pharmacokinetic data of conjugated trastuzumab and total trastuzumab, supporting the inclusion of both proteolytic degradation and decoupling as clearance pathways in the hypothesized metabolic scheme of ado-trastuzumab emtansine. This model attributed the faster clearance rate of conjugated trastuzumab compared to naked trastuzumab to the presence of the decoupling process and indicated that ado-trastuzumab emtansine and trastuzumab have similar proteolytic clearance rates.
Empirical models (e.g., logistic regression, time-to-event) can be used to study the exposure-response relationships for safety and efficacy endpoints.Bone marrow suppression is often associated with ADCs. This toxicity is primarily attributed to the cytotoxic drug component of ADCs. Bender et al. developed a population pharmacokinetics/pharmacodynamics model characterizing the impact of conjugated trastuzumab concentration on platelet counts in HER2-positive metastatic breast cancer patients. Similar models can be developed to describe the time course of ADC effects on other clinical safety endpoints. For example, empirical linear mixed-effects models can be used to study the impact of each ADC analyte on QT/QTc interval prolongation. Additionally, tumor growth inhibition models can be developed to study the effects of ADC analyte exposure and dosing regimens on tumor dynamics.
16. Combining Physiologically-Based Pharmacokinetic and Quantitative Systems Pharmacology Models with Clinical Data
The clinical modeling approaches discussed so far for analyzing clinical data are typically developed from a rich statistical perspective to capture the impact of inter-patient variability on clinical responses and potentially available biomarker data, but often have limited depth in mechanistic details. With the ongoing development of mechanistic PBPK and quantitative systems pharmacology modeling frameworks for ADCs, the application of these methods is likely to expand to gain more insights from clinical data and inform clinical development decisions.
PBPK models have been proposed and applied in preclinical and clinical ADC development. The small molecule components of ADCs are cytotoxic, and the purposes of PBPK modeling for ADCs may include three aspects: (i) accurately predicting the concentrations of free, active cytotoxic drugs in non-target tissues that do not express the target; (ii) predicting the accumulation of ADCs in non-target tissues that may express the antigen; and (iii) simulating the effects of drug-drug interactions (DDIs) that may increase the concentrations of active drugs or affect the concentrations of other drugs. PBPK modeling of ADCs requires extensive data, as the tissue distribution of both the antibody and cytotoxic drug must be characterized. Chen et al. provided an example of such work, parameterizing a minimal PBPK model for the antibody conjugate MMAE and free MMAE. Two different minimal models were developed for ac-MMAE and MMAE using the Simcyp population-based simulator. The ac-MMAE model was limited to the liver, portal vein, systemic circulation, and a single regulating compartment. The single regulating compartment helped align the minimal PBPK model with the observed pharmacokinetic curves. Their MMAE model included only the liver, systemic circulation, and a single regulating compartment. Clinical data were used to inform various aspects parameterizing the ac-MMAE model, including clearance rates, volume of distribution, single regulating compartment volume, and flow rates in and out of the single regulating compartment. Information from clinical, in vitro, and computational sources was used to parameterize the MMAE model. Notably, the hepatic metabolic clearance rate of MMAE via cytochrome P450 3A4 was estimated through extrapolation from in vitro studies. Clinical studies using ketoconazole were used to derive the parameterization of total clearance of MMAE via biliary excretion, combined with human mass balance and rat bile duct cannulation studies. Other parameters, such as the volume of the regulating compartment and the transport rates to the systemic circulation compartment, were estimated using pharmacokinetic simulations with clinical data. For DDI simulations, in vitro data were used to estimate the metabolic clearance rate of MMAE via CYP3A4 and the inhibitory effects of MMAE on CYP3A4.
Research has proposed PBPK frameworks that can simultaneously simulate more aspects of ADCs and their cytotoxic drug components. These frameworks include models that consider distribution effects in both healthy tissues and tumors and cover tumor pharmacodynamics; ADC species with different drug-antibody ratios incorporated into tumor, minimal tissue, and lymphatic compartments, with multi-tissue distribution models for cytotoxic drugs; models that include the tissue transport processes of ADCs and cytotoxic drugs; and frameworks for characterizing tissue distribution data of labeled ADCs and labeled cytotoxic drugs. As more ADC drugs are approved and tissue distribution datasets are improved, it is expected that more parameterization studies and predictive work based on clinical data will emerge, likely driving further clinical development.
QSP (Quantitative Systems Pharmacology) models are typically designed to mechanistically simulate pathways associated with disease processes that are known or presumed to be associated with efficacy endpoints. Therefore, QSP models can serve as a basis for constructing quantitative hypotheses to elucidate the role of specific pathways in treatment intervention responses.For ADC drugs, these efficacy endpoints include pharmacodynamic outcomes such as tumor shrinkage. Several modeling development efforts have integrated pharmacokinetics with tumor stromal delivery, cellular target binding, and downstream steps leading to dynamic changes in tumor size. There is significant potential to merge QSP approaches that simulate ADC efficacy with pathways developed for PBPK models, for example, to assess the potential risks relative to non-target tissue antigen-mediated toxicity endpoints and the impact on tumor shrinkage.
So far, QSP models have primarily been used to explore ADC optimization, preclinical dosing regimen-related issues, and generate translational predictions between preclinical species and humans through mechanistic extrapolation. However, as more clinical data become available, analyzing clinical outcomes and biomarker data using QSP methods will form the basis for the following work: constructing quantitative hypotheses about physiological system states, extrapolating to relevant indications (i.e., tumor types), assessing the impact of new dosing regimens, and exploring the effects of combination therapies on relevant and potentially synergistic pathways. For example, clinical data are often used to constrain and calibrate QSP models of disease pathophysiology and treatment responses at the population level. The strategies, inputs, and steps for QSP model development and application may vary depending on the biological scope being modeled, the scale of the model, and the specific questions being addressed, but there are certain commonalities in the workflow of model development and application. To explore population heterogeneity, feasible boundaries may be established for simulated biomarkers and outcomes based on literature or internal measurement data. When introducing population-level variability, the selection of parameter boundaries and the prioritization of parameters may also be involved. To establish confidence in the model and the selection of unvaried parameters (if any), it may be necessary to develop at least one initial reference virtual patient that meets all constraints and may exhibit additional ideal characteristics, such as an average response to interventions. For example, Shoda et al. conducted extensive validation under multiple intervention conditions for a single non-obese diabetic mouse model. Although various multivariable sensitivity analysis methods can be directly applied to the model, virtual population techniques have been enhanced to explore the sensitivity of QSP models while considering clinical distribution data. Various virtual population algorithms have been developed to accommodate different model scales, parameter search dimensions, data types, expected analysis objectives, and computational resource scenarios. Therefore, it is anticipated that, as has been practiced in many other therapeutic areas, and with the increasing instances of ADC efficacy system model examples, virtual patient populations calibrated against trial results and biomarker data distributions will aid in optimizing protocols, biomarker analyses, and exploring combination therapies in subsequent trial phases. Given the progress made in QSP simulation platforms targeting cancer-immunity cycles, another potential application is screening for immuno-oncology combination regimens that can best enhance ADC responses, integrating the effects of ADC on cell death and antigen presentation with the actions of other pathways such as immune checkpoint inhibition.
17. Summary and Conclusion
This article highlights current thoughts on clinical development strategies for ADCs, from first-in-human dose and regimen selection to the key assessments required to support registration approval. ADCs represent a novel and promising therapeutic platform in oncology. Given their structural composition and pharmacological complexity, clinical pharmacology and modeling and simulation play critical roles in the successful development of such compounds. The field of ADCs is rapidly evolving; therefore, clinical development strategies need to be tailored to specific drugs and remain adaptable as scientific advancements and regulatory insights deepen.
 The public account has been establishedADC Drug Research GroupScan the QR code below to apply for joining. Please provide your name and workplace when joining the group.If you have any questions about ADC drug research, feel free to communicate in the group. Let’s exchange, learn, and progress together.
The public account has been establishedADC Drug Research GroupScan the QR code below to apply for joining. Please provide your name and workplace when joining the group.If you have any questions about ADC drug research, feel free to communicate in the group. Let’s exchange, learn, and progress together.