Skip to content
As of now, regarding thetoxicity of ADCs in solid tumors and preventive measures, the following is known:
① The indications for ADCs are rapidly expanding, gradually shifting from late-stage to early-stage, and from single therapy to combination strategies.
② The toxicity characteristics of most ADCs are similar to the cytotoxicity of their payloads.
③ Some ADCs can also observe unconventional and potentially life-threatening toxicities, necessitating greater understanding of these events and optimization of diagnostic and management practices.
④ The industry is seeking various pharmacological modification strategies to improve the tolerability of ADCs, including molecular changes to the antibody portion, linker, and/or cytotoxic payload.
⑤ Exploring different dosing regimens in randomized trials and researching dose strategies that adapt to responses can optimize the use of ADCs, maximizing their therapeutic value for each indication.
⑥ There are extensive efforts underway to identify toxicity biomarkers in patients receiving ADC treatment and to develop diagnostic tools that can predict and/or detect toxicity early.
Approved ADCs for Solid Tumors: Structure, Major Toxicities, and Interpretations
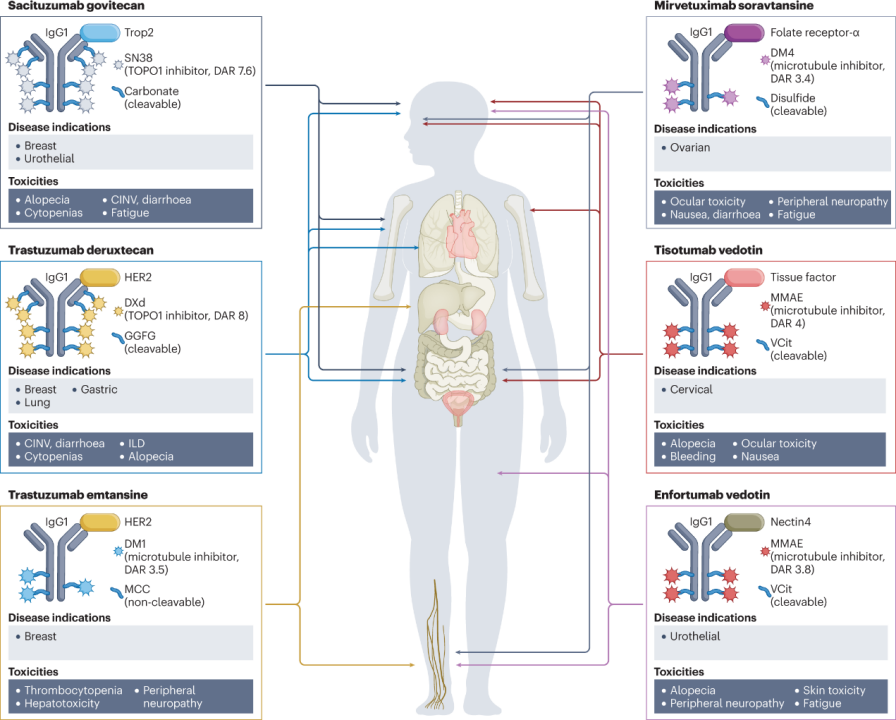
So far, the FDA and EMA have approved six ADC drugs for patients with solid tumors.The following image describes the composition of each ADC (in terms of target antigens, monoclonal antibody types, payloads, and linkers), currently approved indications, and the most common toxicities observed for each drug (as shown in Figure 1 and Table 1). The adverse reaction characteristics of each ADC are usually a mix of on-target and off-target effects, with the latter often determining the maximum tolerated dose. Common adverse reactions observed to varying degrees in many ADCs include fatigue, hair loss, cytopenia, and gastrointestinal disturbances.
Figure 1: Structures and Major Toxicities of Currently Approved ADCs for Solid Tumors
(Figure Legend: CINV, chemotherapy-induced nausea and vomiting; GGFG, Gly-Gly-Phe-Gly; DAR, drug-to-antibody ratio; DXd, deruxtecan; ILD, interstitial lung disease; MCC, maleimide methylcyclohexane-1-carboxylate; MMAE, monomethyl auristatin E; TOPO1, topoisomerase 1; Trop2, trophoblast cell surface antigen 2; VCit, valine-citrulline.)
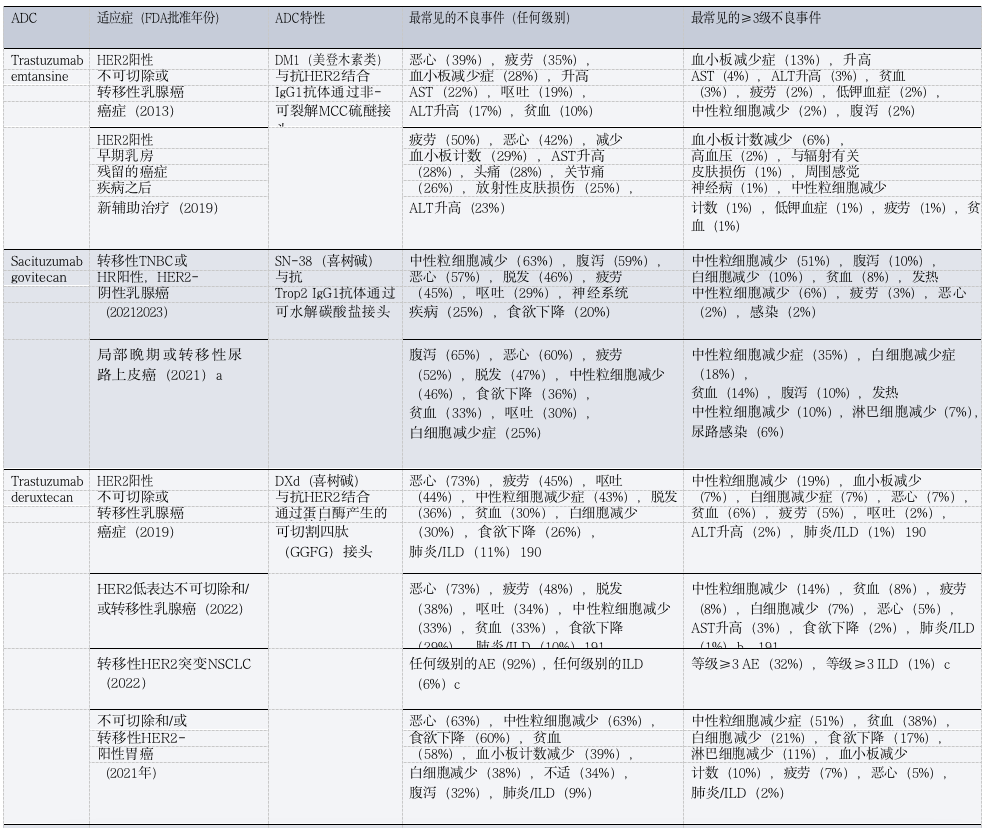
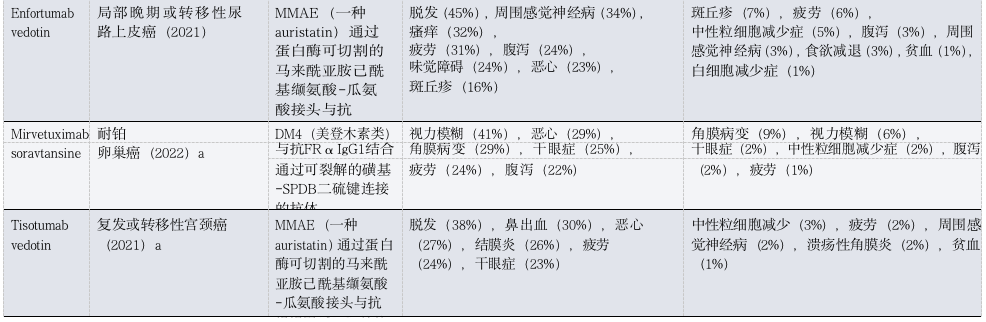
Table 1: Toxicities of FDA-Approved ADCs in Patients with Solid Tumors
(Table Legend: ADC, antibody-drug conjugate; AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; DXd, deruxtecan; FRα, folate receptor-α; GGFG, Gly-Gly-Phe-Gly; HR, hormone receptor; ILD, interstitial lung disease; MCC, maleimide methylcyclohexane-1-carboxylate; MMAE, monomethyl auristatin E; NSCLC, non-small cell lung cancer; TNBC, triple-negative breast cancer; Trop2, trophoblast cell surface antigen 2. a Approved only. b Includes three treatment-related deaths. c. Data is available only in conference abstracts.)
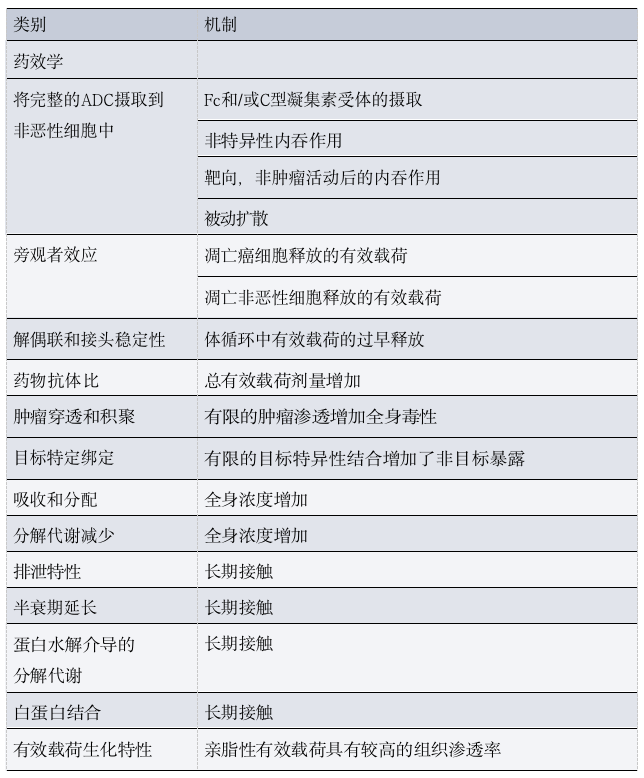
Given the large molecular weight, ADCs are usually administered via intravenous injection.In vivo experimental data suggest that this route of administration may be related to reduced activity and severe skin toxicity for certain ADCs. Ideally, ADCs should remain intact in circulation after injection and release their cytotoxic payload only within or near targeted tumor cells. Minor changes in the structure of ADCs can lead to significant changes in the pharmacokinetic and/or pharmacodynamic characteristics of the drug (Table 2).
After injection, ADCs typically circulate in the bloodstream as a dynamic mixture of intact ADCs (greater than 90% of the composition), unbound drugs (or drug-linkers), and dissociated antibodies. From circulation, ADCs gradually diffuse into the interstitial space of body tissues, ultimately reaching targeted tumor cells, estimated to be about 0.1% of solid tumors.
Once ADCs reach the tumor microenvironment (TME), they are generally believed to bind to target antigens expressed on the surface of cancer cells, undergo internalization in endosomes, and release their payload through chemical or enzymatic cleavage in lysosomes, ultimately leading to necrosis or apoptosis, depending on the mechanism of action of the payload and the concentration achieved at the target site.
More hydrophobic payloads (e.g., monomethyl auristatin E (MMAE) and exatecan derivatives) can diffuse outside the target cells after decoupling from the antibody, thereby exhibiting “bystander killing” effects on antigen-negative cells, which can enhance the antitumor activity of certain ADCs.
This effect also constitutes a determinant of toxicity: the released payload can also enter adjacent non-malignant cells via passive diffusion or transporter-mediated uptake, potentially leading to off-target cytotoxicity.
In addition to the traditionally recognized mechanisms of intracellular targeted payload release in tumor cells, some ADCs can release cytotoxic payloads without antigen involvement and internalization. For example, sacituzumab-govitecan has been shown to release its SN-38 payload extracellularly within the TME, a mechanism that may explain the activity and toxicity of this and other ADCs.
Table 2: Pharmacological Determinants of ADC Toxicity
As mentioned above, most components of ADCs do not reach tumor cells and are gradually degraded and eliminated through a combination of specific and non-specific mechanisms, including target-mediated clearance, Fcγ receptor (FcγR)-mediated uptake, and/or phagocytosis by macrophages located in various tissues.
Importantly, before degradation and excretion, the payload-linker complex released from the ADC using thiol-maleimide chemistry (which constitutes the majority of approved ADCs) can react with free cysteine residues in serum albumin, resulting in long-lived albumin-linker-payload conjugates.
Discussing ADC Toxicity (Starting from Structure)
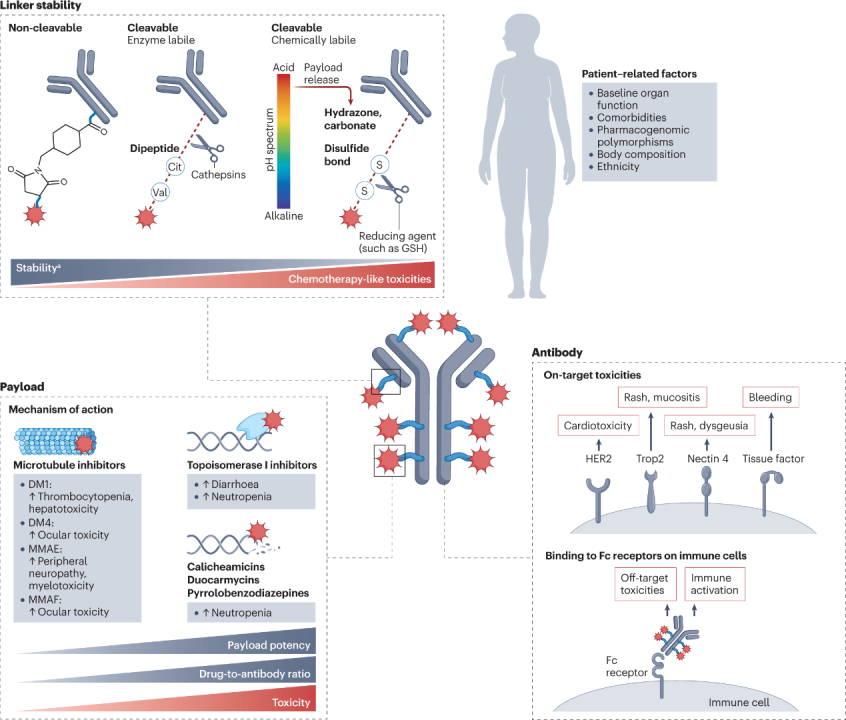
ADCs have a modular structure, and minor modifications to any key component can lead to significant changes in clinical characteristics (Figure 2). This section will analyze the relative contributions of each ADC component and the role of patient characteristics in the types and severity of observed toxicities.
Figure 2: Determinants of ADC Toxicity
(Figure Legend: Cit, citrulline; glutathione; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; Trop2, trophoblast cell surface antigen 2. Highly stable linkers are associated with increased incidence of certain adverse reactions, including ocular toxicity, neurotoxicity, or hepatotoxicity, depending on the specific drug.)
According to the fundamental principles behind ADC development, it is expected that the targeted antigen will determine the toxicity characteristics of the drug. However, clinical experience indicates that most adverse events associated with ADCs share similarities in spectrum, incidence, and severity with the payload backbone, and different ADCs sharing the same payload often exhibit similar toxicity profiles, regardless of differences in target antigens.
These toxicities can be broadly categorized into off-target, off-tumor effects unrelated to the antibody-targeted antigen, and targeted, off-tumor effects arising from the antibody binding to homologous antigens located in non-malignant tissues.
Off-target, extra-tumor toxicity predominates the toxicity profile of most ADCs, typically resulting in adverse reaction characteristics similar to the payload.
The key mechanism of off-target, extra-tumor toxicity of ADCs is believed to be at least partially related to the premature decoupling of the payload in circulation, leading to the diffusion of free cytotoxic payloads into off-tumor compartments. This payload is typically a lipophilic molecule that can permeate cell membranes and enter non-target non-malignant cells.
As mentioned earlier, some payloads may also bind to serum albumin and other thiol-containing circulating plasma proteins, which can increase the half-life of the payload-linker complex and potentially lead to payload deposition in non-malignant tissues.
In addition to the mechanisms by which payloads dissociate from ADCs, other mechanisms are believed to mediate non-malignant cell exposure to cytotoxic payloads, including non-specific endocytosis, endocytosis of intact ADCs in non-malignant cells; and off-target, receptor-mediated uptake caused by interactions between the Fc region of the antibody backbone and Fc receptors expressed by immune cells. The latter mechanism may be more relevant to highly stable ADCs, which are less likely to undergo premature dissociation and release into circulation, thus more likely to encounter intact ADC in non-malignant tissues. Regardless of the mechanism, the extent of non-malignant cell exposure to off-target payload ultimately determines the tolerability of the agent, making the selection of payload a key decision in any ADC design.