Purdue University,Professor Christopher Uyeda’s team utilized the principles of asymmetric catalysis to achieve control over the E/Z stereoisomers in the formation of non-chiral alkene products. This reaction involves cobalt-catalyzed reductive coupling of 1,1-dichloroalkenes with allylic alcohols, resulting in the formation of skipped 1,4-diene products.
Abstract
Catalytic asymmetric reactions are primarily used to install new stereogenic centers highly enriched in either theR or the S configuration. Here, we demonstrate that chiral catalysts can also be used to control the E/Z geometry of an alkene product devoid of chirality. The process is akin to a parallel kinetic resolution in the sense that a chiral catalyst converts two enantiomers of a starting material to two different products, in this case, only differing in the E/Z geometry of an alkene. The reaction is a reductive addition of a 1,1-dichloroalkene to a chiral secondary allylic alcohol. The catalyst controls the facial selectivity of addition to the allylic alcohol, and a subsequent anti-selective β-hydroxide elimination dictates the E/Z geometry of the alkene product. Synthetic applications of the skipped 1,4-diene products and studies probing the origin of stereoselectivity are described.
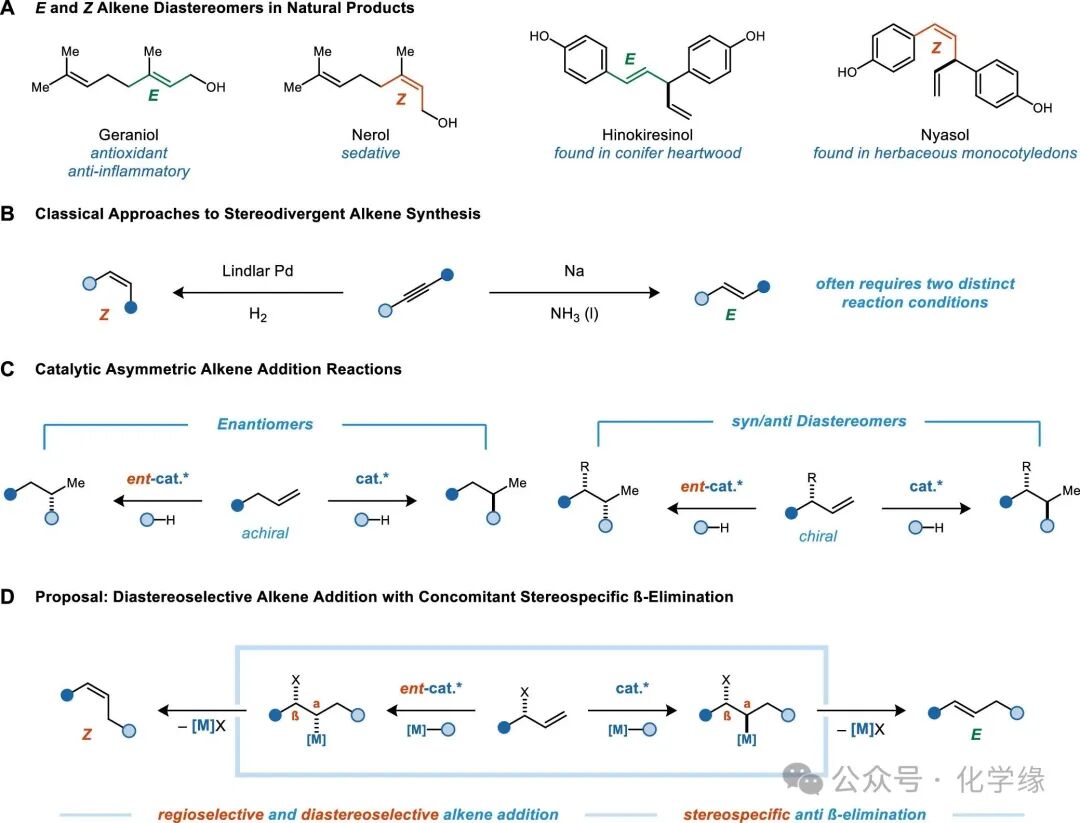
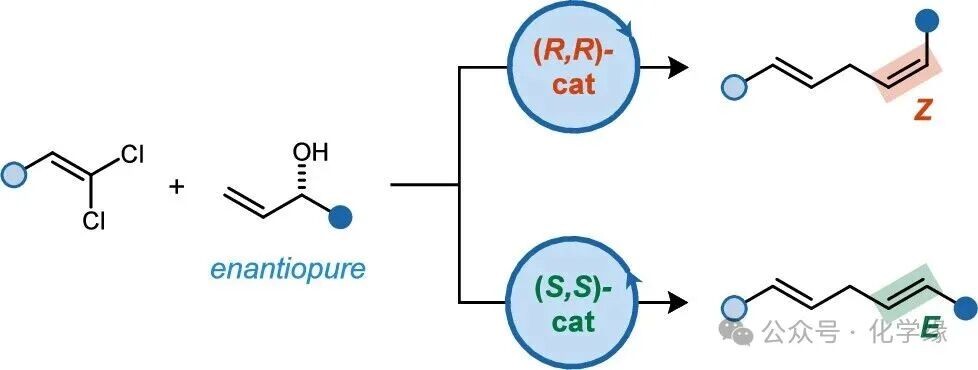
Figure 1 Research background and reaction design
Control over the E/Z configuration is one of the most prominent challenges in the field of alkene synthesis. Traditionally, selective synthesis of E or Z alkenes relies on two distinctly different mechanisms: for example, semi-reduction of alkynes under solvated metal conditions (E selectivity) or hydrogenation reactions catalyzed by transition metals (Z selectivity). In recent years, research has shifted towards exploring stereodivergent catalytic reactions that can simultaneously generate both E and Z alkenes. In most cases, such processes involve Z selective reactions, which are then converted to the thermodynamically more stable E alkenes through isomerization. Only in a few cases can both E and Z alkenes be generated simultaneously through kinetic control. Establishing a general strategy for the synthesis of stereoisomers of E and Z alkenes under catalyst control would avoid the complex separation of alkene enantiomers or the need for alternative synthetic routes to design new pathways for the synthesis of E and Z alkenes.
This article reports that the principles of asymmetric catalysis can be used to achieve control over the E/Z stereoisomers in the formation of non-chiral alkene products. The reaction involves cobalt-catalyzed reductive coupling of 1,1-dichloroalkenes with allylic alcohols, resulting in the formation of skipped 1,4-diene products. In typical catalytic asymmetric alkene addition reactions, chiral catalysts introduce reagents to one of the two prochiral faces of the alkene, resulting in enantiomerically enriched products. If the substrate itself contains a stereocenter with a defined configuration, the reaction will exhibit non-enantiomeric selectivity rather than enantiomeric selectivity. If such non-enantiomeric selective alkene addition is coupled with the stereospecificity of the allylic leaving group in the β-elimination reaction, the E/Z configuration of the resulting alkene will be determined by the enantiomer of the catalyst used.
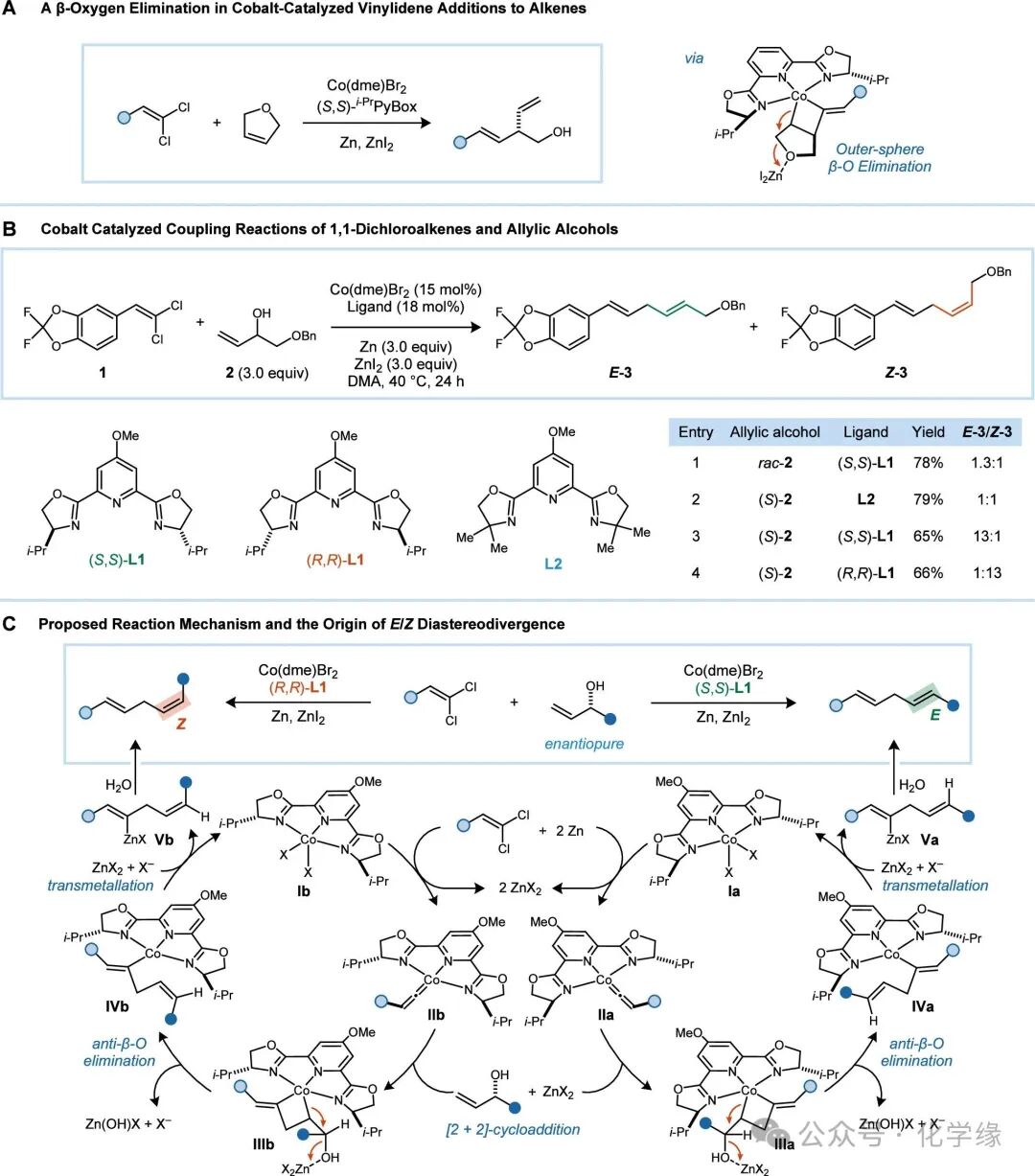
Figure 2 (A) Cobalt-catalyzed 1,1-dichloroalkene reductive addition to 2,5-dihydrofuran showing β-elimination phenomena. (B) Reductive addition of 1,1-dichloroalkenes to allylic alcohols, generating 1,4-diene products. The selectivity of E/Z is determined by the chirality of the allylic alcohol and the catalyst. (C)E/Z stereodivergent reaction mechanism and origin.
The reaction is based on the study of the mechanism of the allylic addition of chiral catalysts to alkenes. When using (PyBox)Co catalysts, the addition of 1,1-dichloroalkenes to Z alkenes generates highly enantiomerically enriched axial chiral methylene cyclopropanes. However, when the substrate contains allylic alcohol salts, no methylene cyclopropane products were observed, but rather high yields of β-alcohol salt elimination products were generated. Despite the lack of direct experimental evidence for the precise mechanism of elimination, DFT models suggest that Lewis acid-assisted trans-elimination may be favored.
We first studied the reductive coupling reaction of 1,1-dichloroalkene 1 with allylic alcohol 2. Using racemic alcohol 2 and the cobalt catalyst loaded with (S,S)-L1, a yield of 78% was obtained for the 1,4-diene product 3, which is a mixture of E and Z enantiomers in a ratio of 1.3:1. In complementary experiments, the use of a non-chiral ligand L2 with (S)-2 (>99% ee) also yielded E/Z of 1:1 mixture 3. It is speculated that E and Z alkenes may form through two enantiomeric pathways, and subsequently, the enantiomerically pure ligand (S,S)-L1 was tested in combination with enantiomerically pure (S)-2. The result yielded 65% yield of product 3, with an E/Z ratio of 13:1. When using the trans ligand (R,R)-L1, the reaction is more inclined to generate Z-3, with an E/Z ratio of 1:13. Thus, it is evident that the reductive alkene addition itself is not inherently E or Z selective, but can achieve high non-enantiomeric selectivity through the use of chiral catalysts.
Based on the observed stereoselectivity of this reaction, the origin mechanism of the isomer induction phenomenon was elucidated. The stepwise reduction of cobalt (II) catalyst and the oxidative addition of 1,1-dichloroalkenes generate cobalt vinyl intermediates II. The subsequent [2+2] cycloaddition reaction with allylic alcohol exhibits regioselectivity, placing the alcohol group at the β position of the Co-C(sp3) bond. The most sterically accessible reaction pathway should be located on the trans side of the vinyl aryl substituent, and in the quadrant unoccupied by the ligand i-Pr group. Cobalt cyclobutane III then undergoes trans-selective β-hydroxide elimination, generating the ethylene cobalt intermediate IV. For the combination of (S,S)-L1/(S)-2, the stereoselectivity of the [2+2] cycloaddition and the β elimination steps jointly determine the generation of E– alkenes, while the combination of (R,R)-L1/(S)-2 generates Z– alkenes. Ultimately, the ethylene cobalt IV undergoes a transmetalation reaction with ZnX2, and the resulting ethylene zinc species quenched by alcohol starting materials or H2O ultimately generates 3.
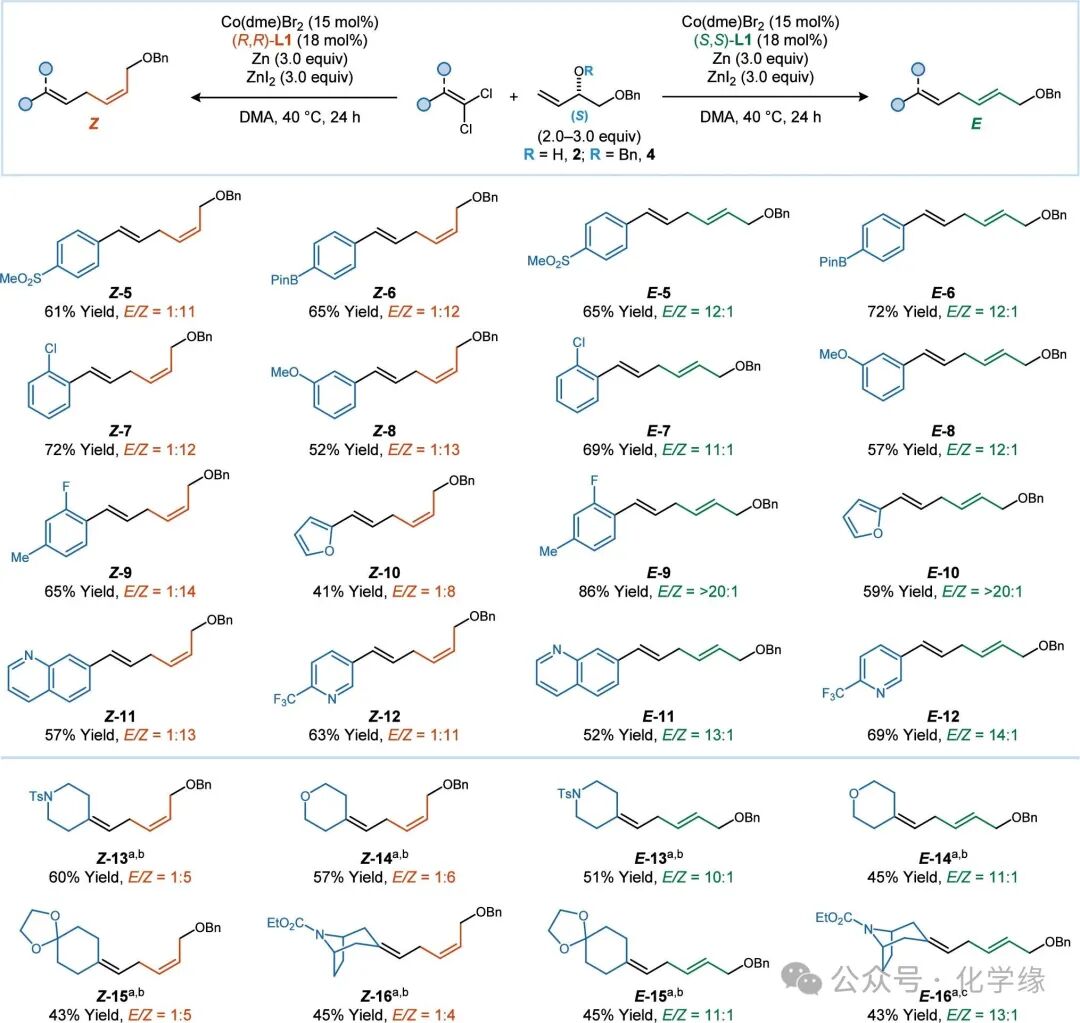
Figure 3 Substrate scope of 1,1-dichloroalkenes
Standard reaction conditions: 1,1-dichloroalkene (1.0 equiv), 2 (2.0–3.0 equiv), Co(dme)Br2 (15 mol %), (S,S)- or (R,R)-L1 (18 mol %), Zn (3.0 equiv), ZnI2 (3.0 equiv), DMA (0.13 M), 40 °C, 24 h.
Summarized the substrate scope of the stereodivergent addition of vinyl and allylic alcohol. Various aryl and heteroaryl substituted 1,1-dichloroalkenes can react effectively. Functional groups commonly tolerated in transition metal-catalyzed cross-coupling reactions, such as boronic esters (6) and aryl chlorides (7), are well tolerated.
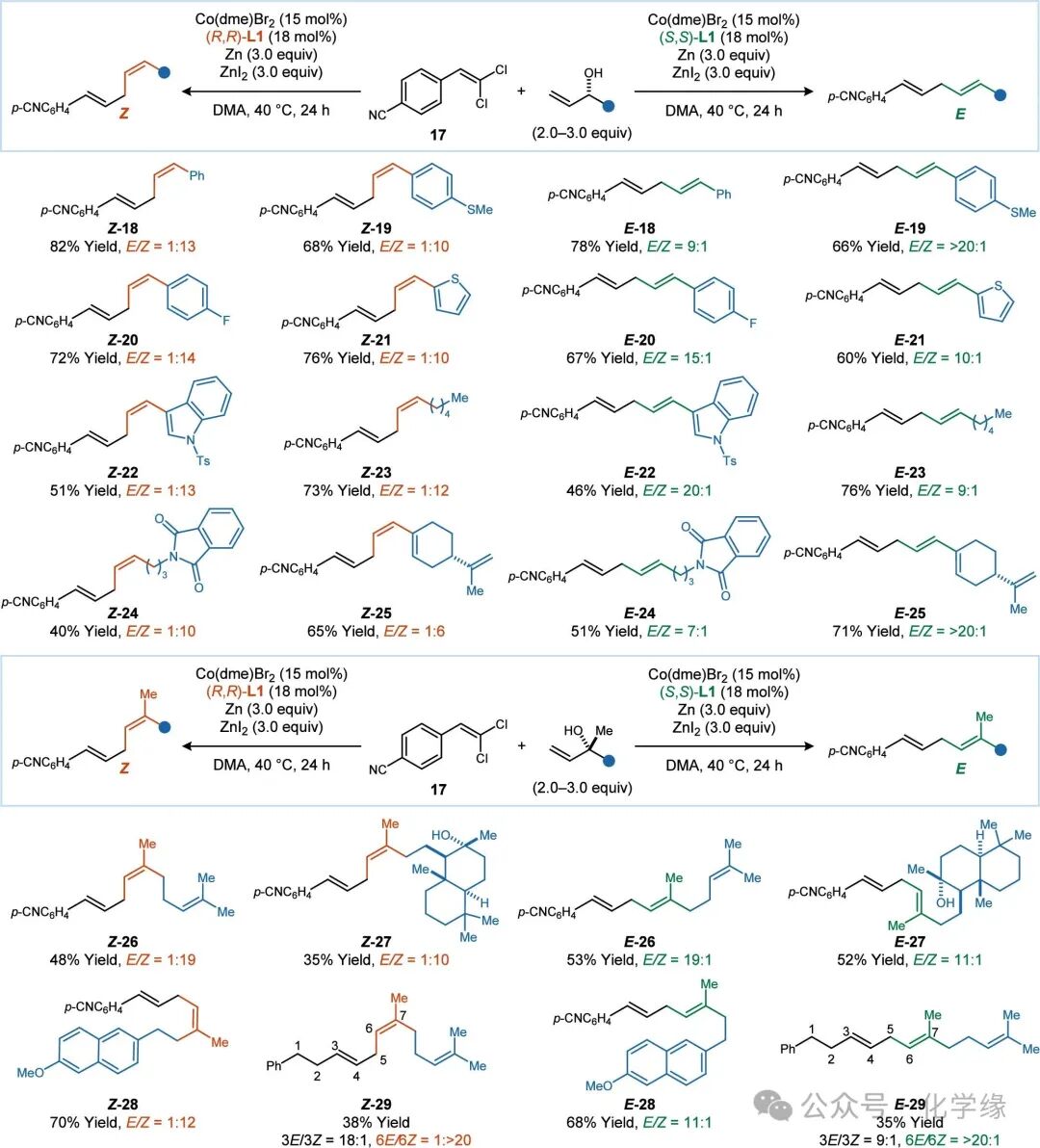
Figure 4 Substrate scope of allylic alcohols
Standard reaction conditions: 17 (1.0 equiv), allylic alcohol (2.0–3.0 equiv), Co(dme)Br2 (15 mol %), (S,S)- or (R,R)-L1 (18 mol %), Zn (3.0 equiv), ZnI2 (3.0 equiv), DMA (0.13 M), 40 °C, 24 h.
When using allylic alcohol 2, fully substituted 1,1-dichloroalkenes did not produce any coupling products. Instead, 1,1-dichloroalkenes were converted to the corresponding monochloroalkenes. For these sterically hindered substrates, the unexpected protonation of cobalt (vinyl) intermediates may dominate the subsequent reduction to vinyl. Therefore, the use of non-acidic allylic benzyl ethers (S)-4 enabled these coupling reactions (products 13–16).
Aryl, heteroaryl, and alkyl substituents can be introduced into the allylic alcohol reactants. For substrates containing multiple alkenes (product 25), terminal alkenes are more reactive than the sterically hindered disubstituted and trisubstituted alkenes. With the many advances in asymmetric synthesis, the types of chiral secondary alcohols used in this reaction can be obtained with high enantiomeric enrichment through various methods: for example, using kinetic resolution of secondary alcohols (8) or through asymmetric addition of allylic nucleophiles to aldehydes (9).
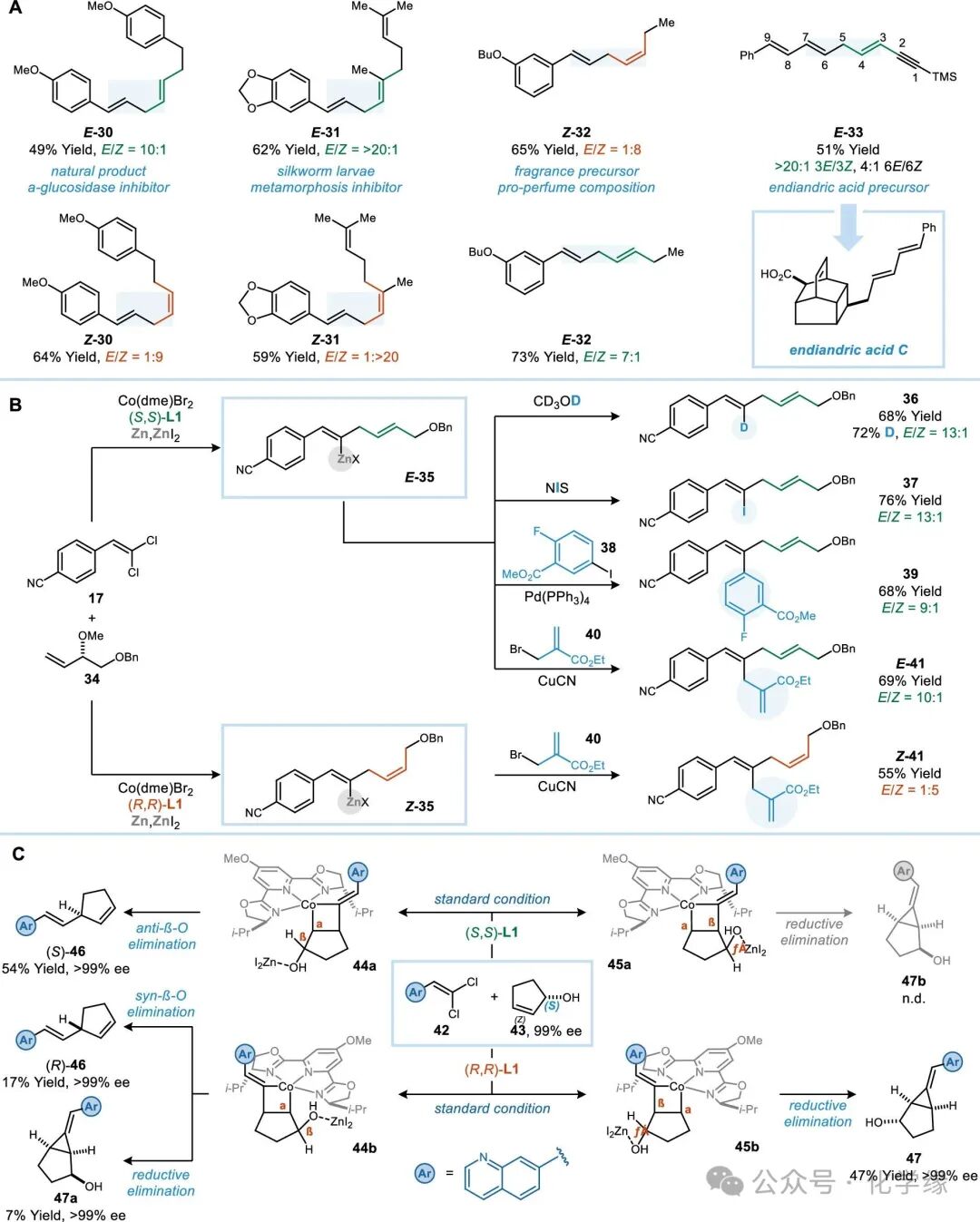
Figure 5 Applications of the reaction
Natural products and bioactive molecules containing chiral tertiary allylic alcohols (such as linalool, anethole, and naproxen) have been shown to be suitable substrates for this reaction, generating products with high selectivity for E/Z isomer selectivity. Compound E-30 was isolated from the plant subgenus Ottelia and exhibits α-glucosidase inhibitory activity. By using the necessary starting materials, both E-30 and its unnatural enantiomer Z-30 can be obtained with high enantiomeric selectivity. The skipped 1,4-diene compound E-30 exhibits inhibitory activity against silkworm larvae. Previously, the synthesis of E-30 through Wittig olefination reactions yielded a mixture of stereoisomers. In contrast, the coupling reaction of vinyl and allylic alcohol allows for the separate synthesis of E-31 and Z-31, achieving enantiomeric selectivity of >20:1.
1,4-diene structures are also present in fragrance compounds and their precursors, which are often derived from terpenoid natural products. Fragrance precursors Z-32 can be efficiently synthesized, and novel analogs E-32 can also be obtained. Finally, 1,4-diene compounds are also used as intermediates in total synthesis. For example, E-33 was utilized in Nicolaou’s synthesis of dicarboxylic acids, yielding a 1:1 E/Z mixture at the C3-C4 alkene position through a nine-step reaction (total yield 35%). Only the E isomer possesses the stereochemical configuration required for the synthesis of A-G type dicarboxylic acids, necessitating separation from the undesired Z isomer via column chromatography. The vinyl coupling reaction at the C3–C4 alkene site achieves an >20:1 E/Z selectivity to obtain E-33 (yield 51%), while at the C6–C7 alkene site, it exhibits a milder 4:1 E/Z selectivity.
To fully utilize the presumed organic zinc intermediates in this reaction, we further prevented their protonation by replacing the free alcohol group in the substrate with a methyl ether. The reductive coupling reaction of 1,1-dichloroalkene 17 with allylic methyl ether 34 generates product 36 with comparable yield and stereoselectivity to the corresponding allylic alcohol. When the crude reaction mixture was quenched with CD3OD, a deuteration rate of 72% was observed at the C2 position, consistent with the proposed structure of the vinyl zinc species. Additionally, quenching with NIS or performing catalytic cross-coupling reactions yielded iodinated products (37), arylation products (39), (14) and allylation products (41). The corresponding Z-selective reactions are also feasible, yielding Z-41. Thus, reductive coupling provides a simplified synthetic route to stereochemically defined trisubstituted dienes and trienes, which are challenging to prepare using other alkene synthesis methods.
To fully utilize the presumed organic zinc intermediates in this reaction, we further prevented their protonation by replacing the free alcohol with a methyl ether. The reductive coupling reaction of 1,1-dichloroalkene 17 with allylic methyl ether 34 generates product 36 with comparable yield and stereoselectivity to the corresponding allylic alcohol. When the crude reaction mixture was quenched with CD3OD, a deuteration rate of 72% was observed at the C2 position, consistent with the proposed structure of the vinyl zinc species. Additionally, quenching with NIS or performing catalytic cross-coupling reactions yielded iodinated products (37), arylation products (39), and allylation products (41). The corresponding Z-selective reactions are also feasible, yielding Z-41. Thus, reductive coupling provides a simplified synthetic route to stereochemically defined trisubstituted dienes and trienes, which are challenging to prepare using other alkene synthesis methods.
The stereoselectivity of the vinyl coupling reaction ultimately depends on the trans selectivity of the alkene addition step and the neighboring/remote selectivity of the β-elimination step. The reaction of 1,1-dichloroalkene 42 with (S)-cyclopent-2-en-1-ol (43) was examined. When using (S,S)-L1, two [2+2] cycloaddition transition states exist: one allows the alkene to approach the Co=C=CR intermediate in the open quadrant (44a and 45a). The intermediate 45a is difficult to form due to steric hindrance, and the corresponding product 47b was not observed. In contrast, the chiral diene (S)-46 was generated with a yield of 54% and >99% ee, indicating that the [2+2] cycloaddition step has extremely high trans configuration selectivity. When using (R,R)-L1, the steric hindrance between the hydroxyl and the cobalt vinyl aryl substituent is weaker, allowing both [2+2] cycloaddition transition states to be accessible energetically. The formation of 45b in the [2+2] cycloaddition transition state places the hydroxyl at the γ position relative to the Co-C(sp3) bond. Therefore, the β elimination reaction cannot occur, ultimately generating methyl cyclopropane 47 with a yield of 47% and >99% ee. In another metal cyclization compound 44b, although the hydroxyl is in a suitable β-elimination position, it is adjacent to the Co-C(sp3) bond. The neighboring β-elimination competes with the C-C reductive elimination pathway, yielding products (R)-46 and 47a at 17% and 7% yields, respectively, both with >99% ee values.
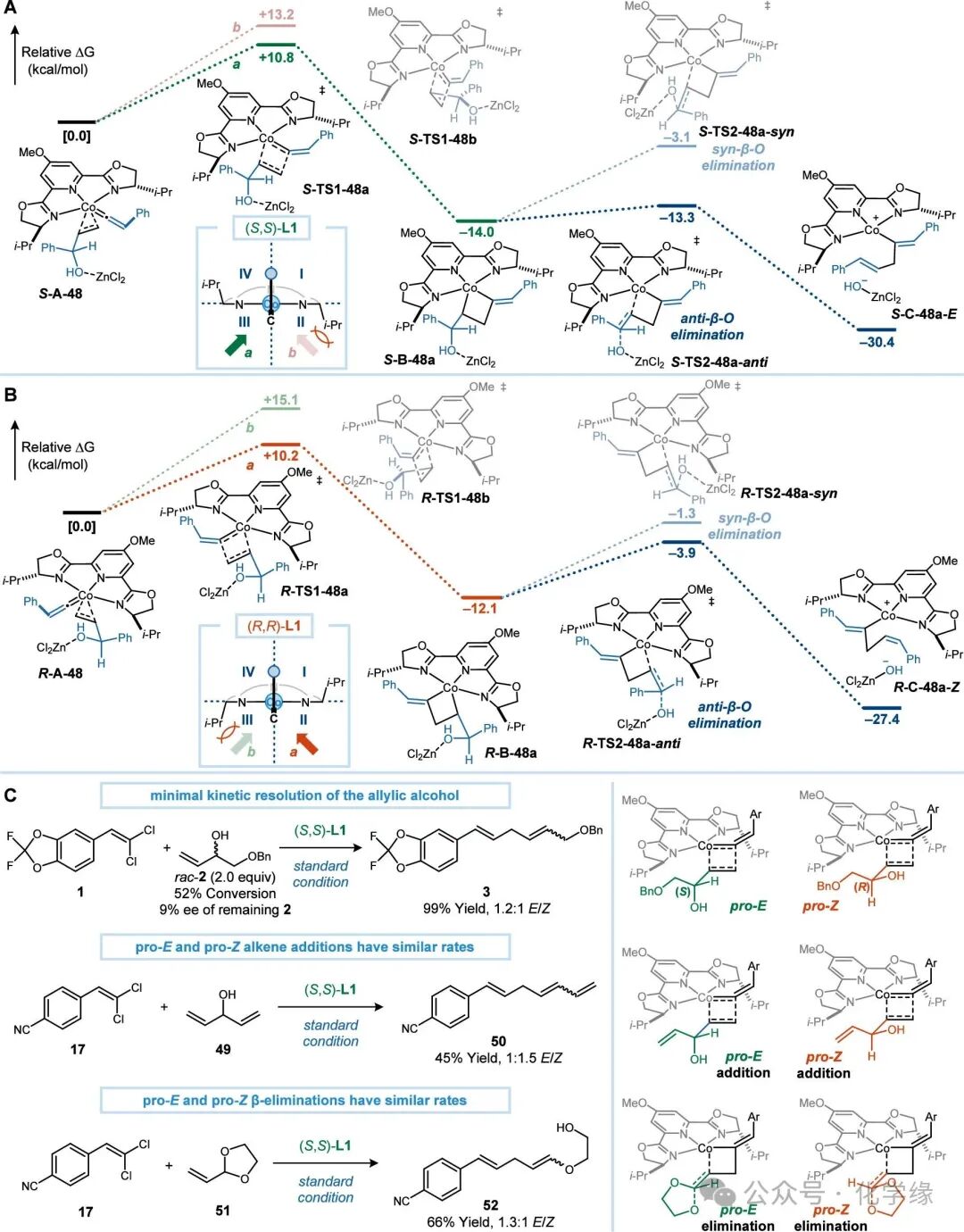
Figure 6 DFT computational results
Based on these results, a DFT model describing the enantiomeric selectivity was established. Using (S,S)-L1 as the model system, the optimal [2+2] cycloaddition transition state of the Co=C=CR intermediate involves approaching in the least sterically hindered quadrant, away from the vinyl substituent and the ligand i-Pr group. Additionally, the hydroxyl is positioned at the β position of the Co-C(sp3) bond, facilitating the subsequent elimination reaction. When the hydroxyl is at the γ position of the Co–C(sp3) bond, the regioisomeric transition state is energetically unfavorable due to steric interactions between the ligand i-Pr group and the coordinated alcohol, which is 9.1 kcal/mol higher in energy.
The metal ring S-B-48a undergoes a near-zero energy barrier (0.7 kcal/mol) transβ elimination reaction, generating E alkene products. The alternative cis β elimination reaction generates Z alkene products, which has a higher barrier of 10.9 kcal/mol. For (R,R)-L1, the [2+2] cycloaddition step generates enantiomeric metal rings R-B-48a. The trans β-elimination is favored due to a smaller energy difference of 2.6 kcal/mol compared to the cis β-elimination, making it easier to form Z alkene products. The catalyst (R,R)- exhibits a higher energy barrier in the β- elimination reaction, possibly due to the need to access a higher energy rotamer to present the hydroxyl in a trans orientation.
DFT models indicate that both pro-E and pro-Z β-elimination reactions have lower activation energy barriers. To experimentally validate this step, vinyl-substituted cyclic acetals 51 were used in the reaction. Using (S,S)-L1 as the reagent, a yield of 66% was obtained for the vinyl ether product 52, with an E/Z molar ratio of 1.3:1. Therefore, in the [2+2] cycloaddition step and after the formation of cobalt ring intermediates, the pro-E and pro-Z β-elimination transition states show no inherent preference.
Conclusion: The asymmetric reductive vinyl and allylic alcohol coupling reaction demonstrates that chiral catalysts can control asymmetric selectivity even in the formation of non-chiral products. While most asymmetric addition reactions targeting chiral substrates form new stereocenters through stereoisomeric selective processes, the uniqueness of this reaction lies in the elimination of both stereocenters to generate alkenes. By using two different enantiomers of catalysts, stereodivergent access to either E or Z alkenes can be achieved with high selectivity. DFT models indicate that the E/Z selectivity depends on both the alkene face selectivity in the [2+2] cycloaddition step and the preference of trans β-elimination over cis β-elimination.
Article Information:
Application of Asymmetric Catalysis in the E/Z-Stereodivergent Synthesis of Alkenes
Mingxin Liu, Vibha V. Kanale, Christopher Uyeda*
DOI: 10.1021/jacs.5c15281
We hope this article inspires you, and let us work together for a better future!
