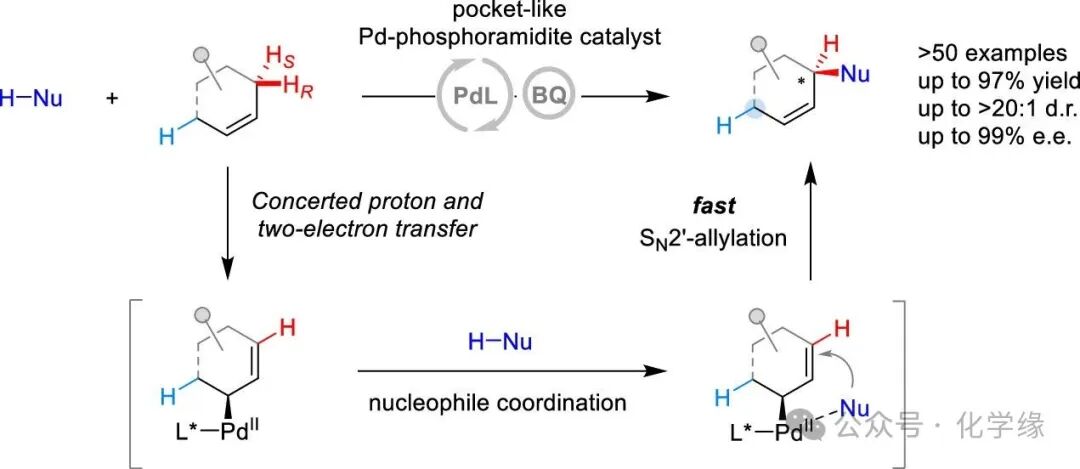
Professor Liu-Zhu Gong’s team at the University of Science and Technology of China reported a site- and enantioselective allylic C–H alkylation of internal alkenes catalyzed by pocket-like chiral phosphoramidite-Pd. This method is applicable to a wide range of cyclic and acyclic internal alkenes.
Abstract
The site- and enantioselective transformation of allylic C-H bonds in ubiquitous internal alkenes represents a significant challenge in asymmetric synthesis. Introducing an enzyme-like chiral environment is a crucial strategy to achieve pro-R/S and site-differentiation of allylic C-H bonds, thereby circumventing the formation of complex regio- and stereoisomeric mixtures. In this work, we report a highly site- and enantiodifferentiating allylic C-H alkylation of internal alkenes through pocket-like chiral phosphoramidite-Pd catalysis. This method is effective for a wide spectrum of cyclic and acyclic internal alkenes. Dispersion interactions are highlighted as playing a pivotal role in stabilizing the enantiodifferentiating transition states of allylic C-H bond cleavage. Within an open pocket delineated by the segments of bulky phosphoramidite ligand, electronically and sterically similar allylic C-H bonds are discriminated. This process culminates in the generation of chiral σ-allylpalladium intermediates that rapidly engage in SN2′-allylation, installing the functional group precisely at the position of the initially cleaved prochiral hydrogen.
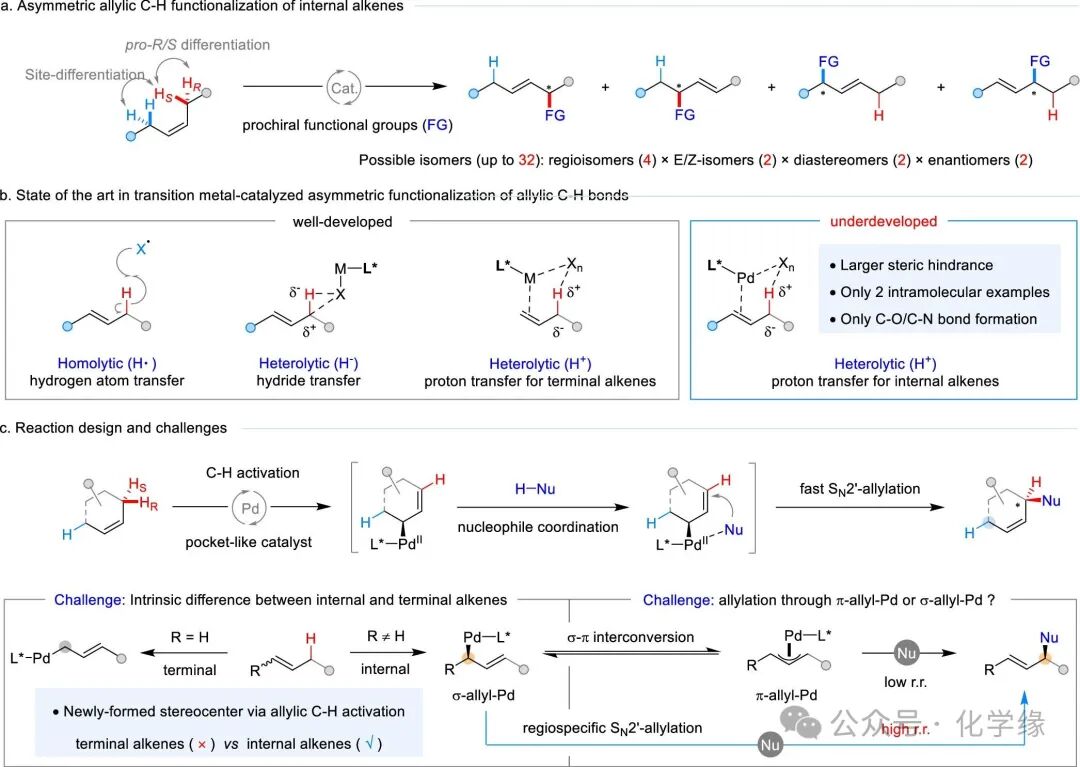
Figure 1 Asymmetric allylic C–H functionalization of internal alkenes
By simultaneously controlling site selectivity and stereoselectivity, the precise optimization of C-H bond synthesis pathways has been greatly enhanced. However, the inherent similarity among various C-H bonds presents significant challenges, necessitating ongoing efforts to propose innovative and effective solutions. A classic problem in this regard is the site-specific and stereospecific transformation of allylic C-H bonds in widely available internal alkenes, aiming to produce enantiomerically enriched building blocks from petrochemical feedstocks while retaining alkenes for further studies. The presence of two sets of electronically and spatially similar allylic C-H bonds complicates the selective allylic C-H activation process. Therefore, precise control over pro-R/S and site differentiation is essential to suppress the formation of complex regioisomeric and stereoisomeric mixtures. For prochiral functional groups, the potential for isomer formation can soar to 32, highlighting the complex challenges of precisely controlling regio-, E/Z, stereoselectivity, and enantioselectivity in these transformations.
The cleavage of allylic C-H bonds in internal alkenes typically relies on two fundamental concepts: homolytic activation achieved through hydrogen atom transfer (HAT) and heterogeneous hydrolytic activation facilitated by transition metal catalysts. The former relies on the selective interception of allylic radicals by chiral metal catalysts, thereby initiating subsequent asymmetric transformations. Notable examples include copper-catalyzed oxidation and cyanation; chromium-catalyzed carbonyl alkenes; and palladium-catalyzed amination. The latter utilizes transition metal-mediated C-H insertion reactions, proceeding through a three-center hydride transfer transition state. In this category, α-arylated carbonyl compounds are widely applied, with a notable example recently in the total synthesis of natural products. However, proton transfer for site- and stereoselective allylic C-H cleavage in internal alkenes, capable of generating electrophilic alkenylmetal intermediates for various asymmetric transformations, remains a long-standing and unresolved challenge. Despite the fact that the oxidative synthesis of alkenyl palladium complexes via 2-methylpropene and PdCl2 has been established for over 60 years, the asymmetric allylic C-H functionalization catalyzed by transition metals has primarily been limited to terminal alkenes, with limited chiral ligands, a field pioneered by Trost, Gong, White, and others. The allylic C-H alkylation of internal alkenes catalyzed by palladium has not been reported, although only two examples of intramolecular variant formation of carbon-heteroatom bonds have been reported by Sasai and Matsunage.
Compared to terminal alkenes, internal alkenes exhibit reduced reactivity towards allylic C-H activation, primarily due to steric hindrance when coordinating with palladium catalysts. Furthermore, the activation of allylic C-H bonds at the alkene position generates a stereocenter, whereas terminal alkenes do not produce such stereocenters. The σ-π conversion of alkenyl palladium is typically very rapid, forming 1,3-disubstituted alkenyl palladium intermediates from internal alkenes, thereby introducing lower regioselectivity ratios in subsequent nucleophilic attack steps. Based on previous successful activations of structurally diverse terminal alkenes’ allylic C-H bonds through cooperative proton and two-electron transfer pathways, the dissociation of allylic C-H bonds in internal alkenes using a similar mechanism, facilitated by pocket-like chiral phosphoramidite ligands, may achieve precise site and enantioselective control of allylic C-H activation. Based on previous findings, nucleophilic coordination can accelerate C–C bond formation through rapid SN2′-allylation pathways. This method is envisioned to enable the rapid conversion of chiral σ-allylpalladium intermediates, providing regioselective products with high regioselectivity. In this case, the functional group will be precisely introduced at the initially cleaved prochiral hydrogen position, thereby achieving site and enantioselective allylic C-H alkylation of internal alkenes through pocket-like chiral phosphoramidite-Pd catalysis.
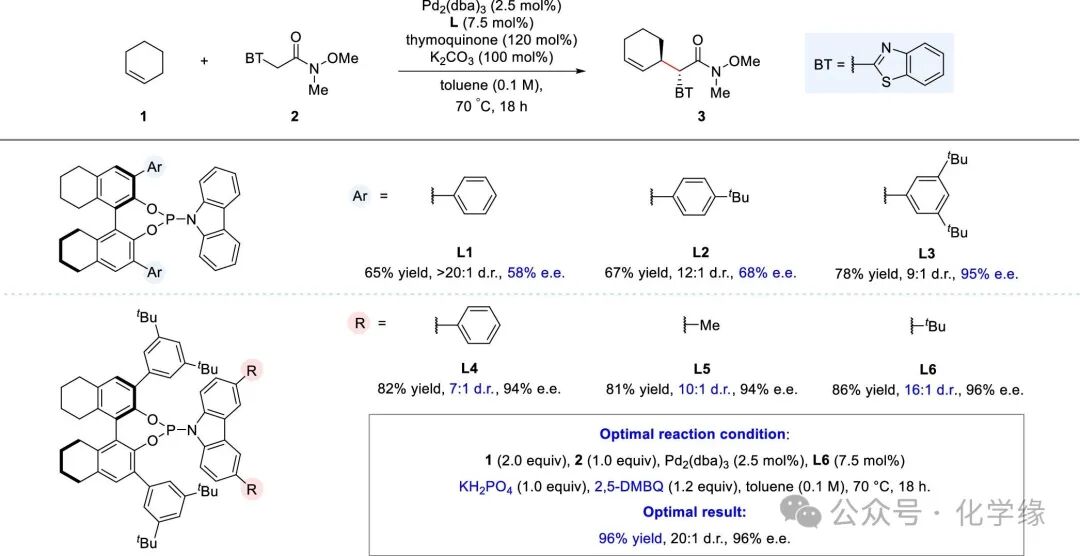
Figure 2 Optimization of reaction conditions
Using cyclohexene1 and α-benzenesulfonamide2 as template substrates for reaction condition optimization, while using Pd2(dba)3 and chiral phosphoramidite ligand L1 as catalysts, thymoquinone as oxidant, K2CO3 as base, and toluene as solvent, at 70°C, the desired alkylation product 3 was obtained with a yield of 65%, >20:1 d.r., 58% ee. After fine-tuning the 3,3′-disubstituted H8-BINOL, it was found that introducing a tert-butyl group at the phenyl position significantly enhanced enantioselectivity (L1 vs L2–L3), although at the cost of reduced diastereoselectivity. By adding a larger 3,5-di-tert-butyl group (L3), the enantioselectivity could be improved to 95%. Additionally, by placing substituents at the 3,6 positions of the carbazole framework, effective recovery of diminished diastereoselectivity while maintaining excellent enantioselectivity can be achieved. Among the various substituents tested, including phenyl and methyl (L4–L5), and tert-butyl (L6), exhibited superior control in non-stereoselectivity. Further refinement of the reaction conditions, using KH2PO4 as base, 2,5-DMBQ as oxidant, yielded the best results with a yield of 96%, 20:1 d.r., 96% e.e.
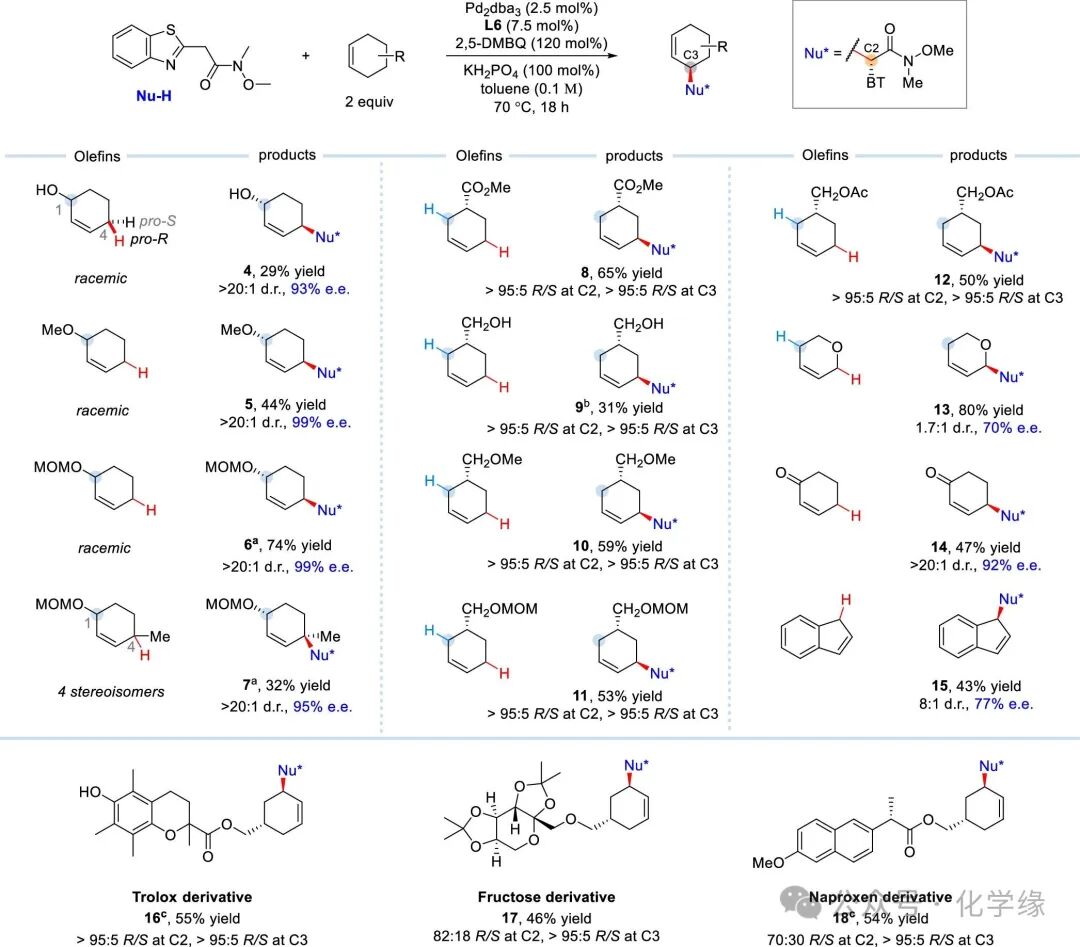
Figure 3 Substrate scope of cyclic internal alkenes
Reaction conditions: alkene (2 equiv), Weinreb amide (1.0 equiv), Pd2(dba)3 (2.5 mol %), L6(7.5 mol %), 2,5-DMBQ (120 mol %), KH2PO4 (100 mol %) and toluene (0.1 M), 70 °C, 18 h
After obtaining optimized conditions, the substrate scope of various asymmetric cyclic alkenes was examined. The results indicate that this pocket-like chiral phosphoramidite–Pd catalyst exhibits enzyme-like chiral recognition of allylic enantiomers (4–6). For these racemically substituted cyclohexenes, the cis hydrogen atom at the 4 position of the R configuration was selectively functionalized, yielding the corresponding alkylation products, >20:1 d.r. 93–99% e.e.. Notably, the presence of ether protecting groups on the hydroxyl group (4 vs 5–6) contributes to improved reaction yields. The S-type exhibited negligible reactivity under the same reaction conditions. In the case of 1,4-disubstituted cyclohexenes (composed of 4 stereoisomers), only the 1R,4S configuration was selectively transformed at the 4 position adjacent to the methyl group, yielding the desired product (7), >20:1 d.r. 95% e.e., although the yield was relatively low. Turning to allylic substituted cyclohexenes, the R configuration alkenes underwent site- and enantiodifferentiating C-H alkylation at the distal alkyl position, producing enantiomerically enriched alkylation products with excellent stereoselectivity (8–12). Additionally, 3,6-dihydro-2H-pyran participated in the allylic C-H alkylation reaction (13), which underwent regioselective modification of the allylic C-H bond adjacent to the oxygen atom. Cyclohexenone, as an electron-deficient alkene, is typically a challenging substrate for palladium-catalyzed allylic C-H functionalization reactions, yet this enantioselective alkylation reaction was successfully accomplished, ultimately yielding the corresponding product (14), >20:1 d.r. 92% e.e.. The five-membered ring indene also exhibited tolerance in this chiral differentiating allylic C-H alkylation reaction, generating the target product (15), although its cis-trans isomer selectivity and enantioselectivity were somewhat reduced. Furthermore, this method is applicable to various internal alkenes conjugated with pharmacologically important molecules, including trolox (16), fructose (17), and naproxen (18), yielding functionalized derivatives with acceptable synthetic results.
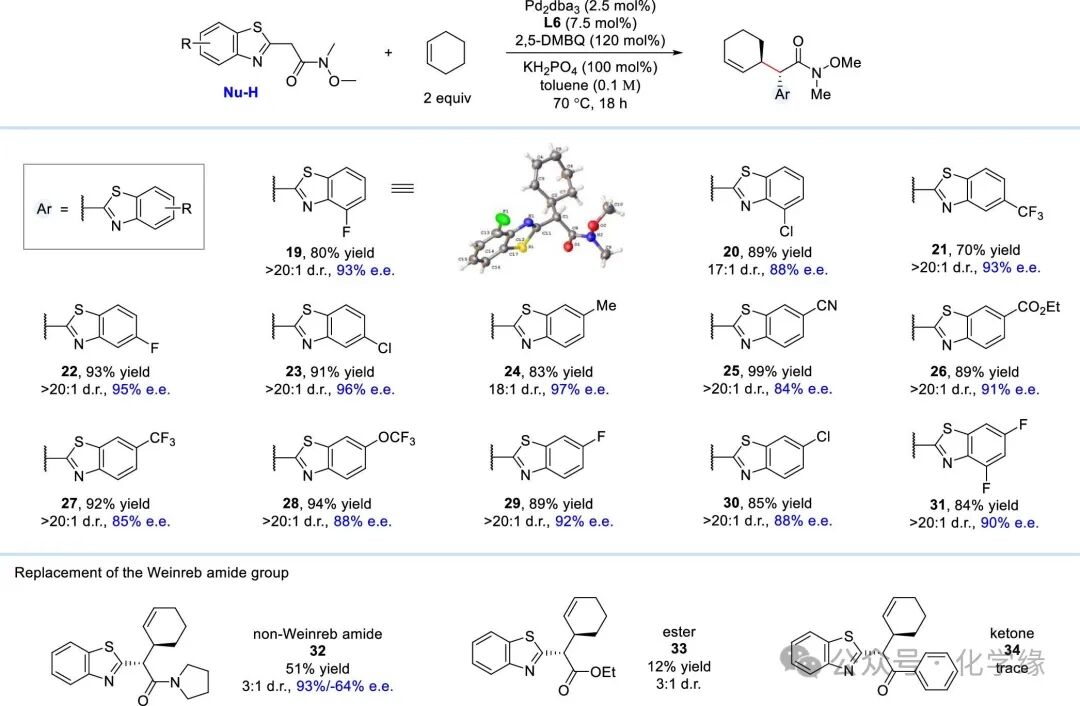
Figure 4 Substrate scope of nucleophiles containing cyclohexene
Reaction conditions: alkene (2 equiv), Weinreb amide (1.0 equiv), Pd2(dba)3 (2.5 mol %), L6(7.5 mol %), 2,5-DMBQ (120 mol %), KH2PO4 (100 mol %) and toluene (0.1 M), 70 °C, 18 h.
Subsequently, the substrate scope of coordinated nucleophiles was explored. At the 4, 5, or 6 positions of the benzenesulfonyl group, whether electron-withdrawing or electron-donating groups, were compatible with the site- and enantiodifferentiating C-H alkylation reaction, yielding the desired products with good to excellent levels of stereoselectivity (19–31). In contrast, replacing the Weinreb amide with simple amides, esters, or ketones weakened reactivity and non-diastereoselectivity (32–34).
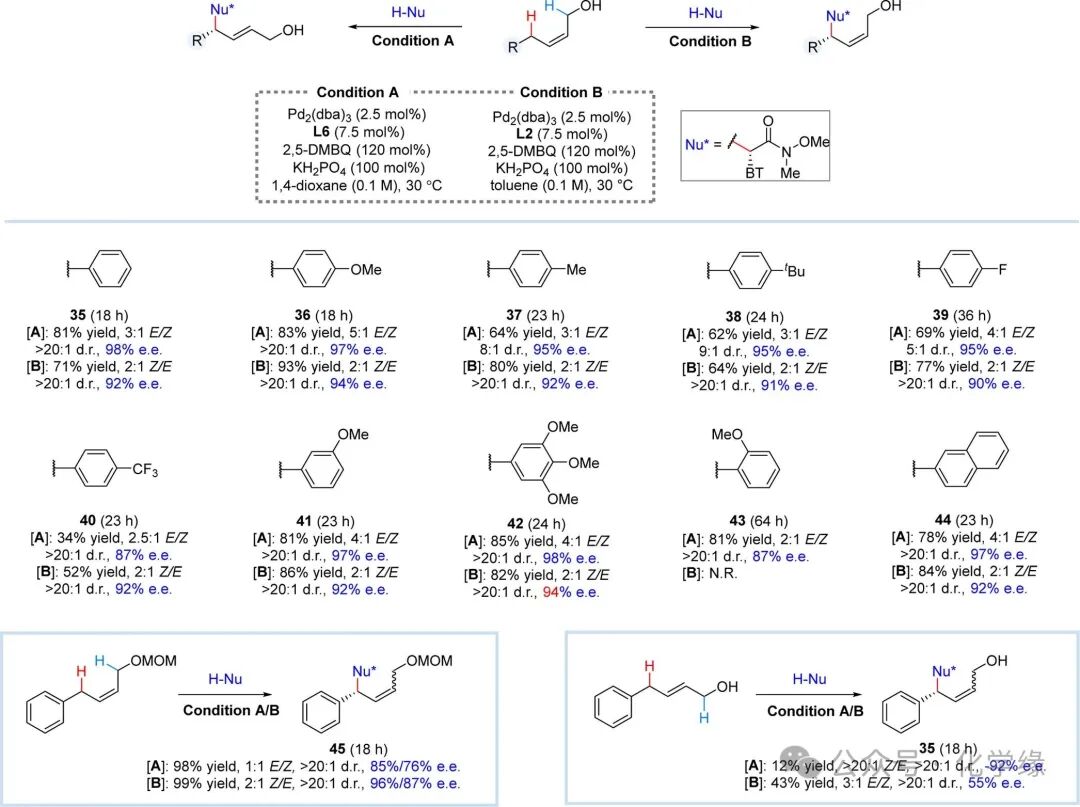
Figure 5 Substrate scope of acyclic internal alkenes
Reaction conditions: alkene (2.0 equiv), Weinreb amide (1.0 equiv), Pd2(dba)3 (2.5 mol %), ligand (7.5 mol %), 2,5-DMBQ (120 mol %), KH2PO4 (100 mol %) and solvent (0.10 M) at 30 °C. Condition A: L6 (7.5 mol %), 1,4-dioxane (0.1 M); Condition B: L2 (7.5 mol %), toluene (0.1 M).
Moreover, the pocket-like chiral phosphoramidite-Pd catalysis also demonstrated adaptability for site- and enantioselective allylic C-H alkylation reactions of non-cyclic internal Z-alkenes. Modifications to the phosphoramidite ligands facilitated a divergent pathway, producing products with excellent non-stereoselectivity and enantioselectivity in E or Z stereochemical configurations. For instance, using L6 as a ligand yielded E-stereochemical alkylation products from (Z)-4-phenyl-2-butanol, while employing L2 favored the formation of Z-stereochemical products, although E/Z selectivity was moderate. The multifunctionality of this E/Z divergent synthesis strategy is exemplified by its successful application in various (Z)-4-phenyl-2-butanols. This method cleverly accommodates both electron-donating and electron-withdrawing substituents at the para position of the phenyl ring, producing E or Z stereochemical products (35–43, with moderate to good yields, high stereoselectivity, and moderate E/Z selectivity. Notably, ortho-methoxy substrates were found to be unreactive under Condition B (43), presumably due to significant steric hindrance in the cleavage of the allylic C-H bond. The compatibility of this method is further demonstrated by its tolerance towards naphthalene rings (44. Additionally, the presence of a methoxymethyl (MOM) protecting group on the hydroxyl group still yielded excellent products (45), although with slightly reduced E/Z and enantioselectivity under Condition A. Furthermore, (E) 4-phenyl-2-butanol was also found to participate in the site-selective allylic C-H alkylation reaction, but the E/Z selectivity was opposite to that of (Z)-4-phenyl-2-butanol. The use of L6 primarily yielded products with high levels of E/Z and enantioselectivity in Z-stereochemical configurations (Z35. The use of L2 with (Z) 4-phenyl-2-butanol yielded E-stereochemical alkylation products (E-35), but with significantly reduced enantioselectivity.
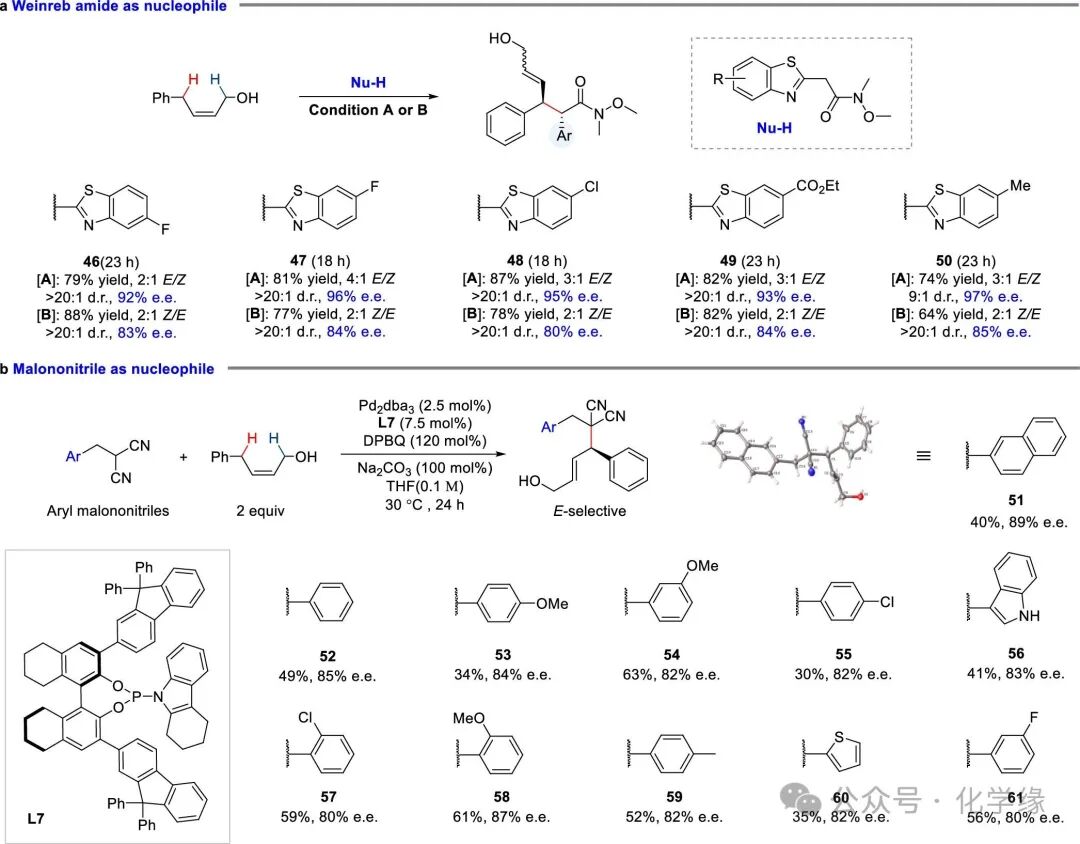
Figure 6 Substrate scope of nucleophiles with non-cyclic internal alkenes
Reaction conditions with Weinreb amide: alkene (2.0 equiv), Weinreb amide (1.0 equiv), Pd2(dba)3 (2.5 mol %), ligand (7.5 mol %), 2,5-DMBQ (120 mol %), KH2PO4 (100 mol %) and solvent (0.10 M) at 30 °C. Condition A: L6 (7.5 mol %), 1,4-dioxane (0.1 M); Condition B: L2 (7.5 mol %), toluene (0.1 M). Reaction conditions with malononitrile: alkene (2.0 equiv), malononitrile (1.0 equiv), Pd2(dba)3 (2.5 mol %), L7 (7.5 mol %), 2,5-diphenyl-1,4-benzoquinone (120 mol %), Na2CO3 (100 mol %) and tetrahydrofuran (0.10 M) at 30 °C.
Regarding non-cyclic internal Z-alkenes, the scope of this method has been extended to substituted carbon nucleophiles. Electron-donating or electron-withdrawing substituents at the 5 or 6 positions of the benzenesulfonyl group exhibited good tolerance, yielding the desired products (46–50). Notably, malononitriles were also found to be compatible, yielding E-type products. Through extensive ligand screening, the sterically demanding L7, bearing a 9,9-diphenyl-9H-fluoren-2-yl group at the 3,3′ position, and utilizing a 1,2,3,4-tetrahydrocarbazole framework, was identified as the optimal solution. Under these conditions, a series of malononitriles underwent asymmetric allylic C-H alkylation, producing E-stereochemical adducts (51–61, with moderate yields (up to 63%), and good to high enantioselectivity (89% e.e).
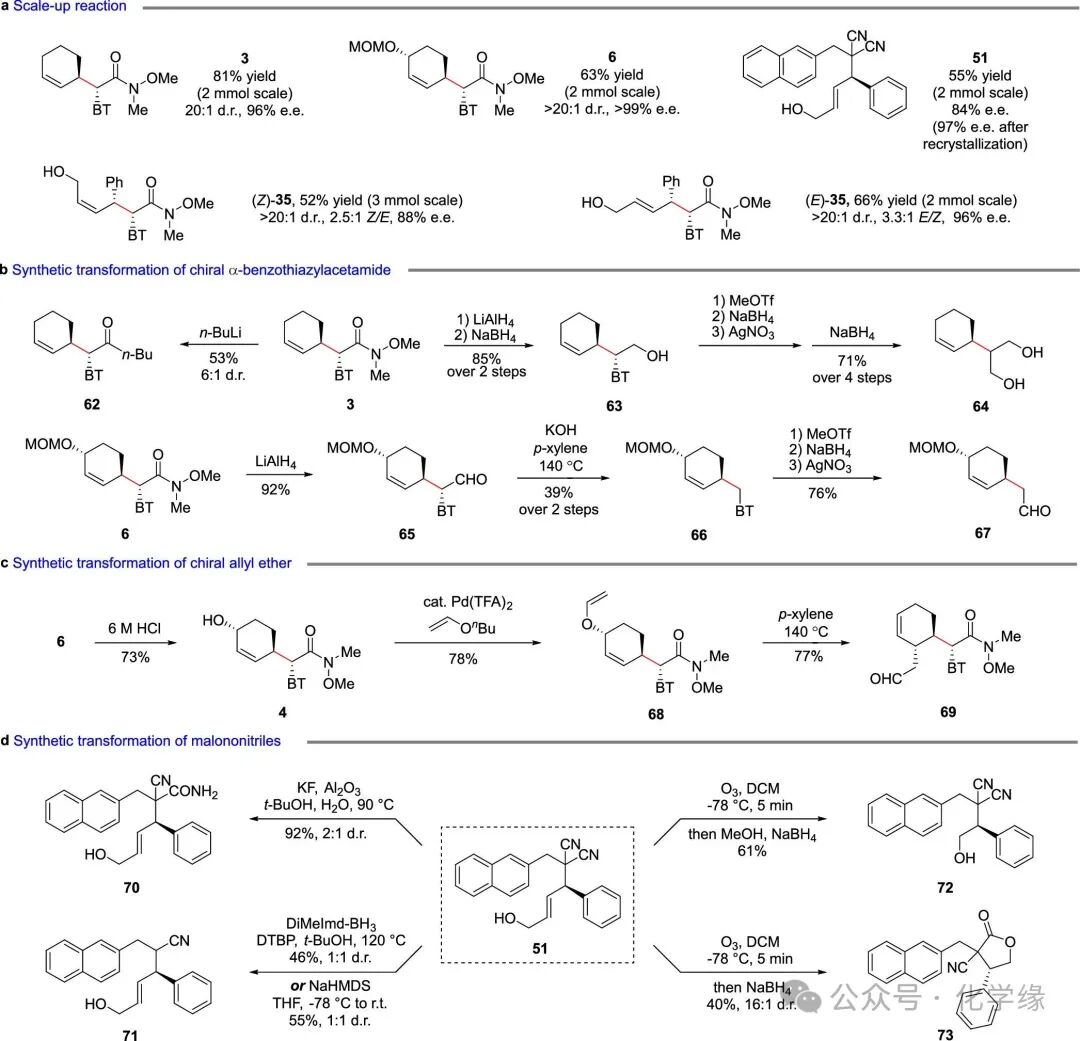
Figure 7 Synthetic applications
To validate the feasibility of the method, gram-scale reactions of cyclic and acyclic internal alkenes using Weinreb amides as nucleophiles produced products 3, 6, and 35, with high yields and maintained stereoselectivity.Weinreb amide 3 underwent nucleophilic substitution with n-BuLi, generating ketone 62 with a yield of 53%, d.r. ratio of 6:1. Sequential reductions using LiAlH4 and NaBH4 yielded 3 into 63, achieving a yield of 85%.
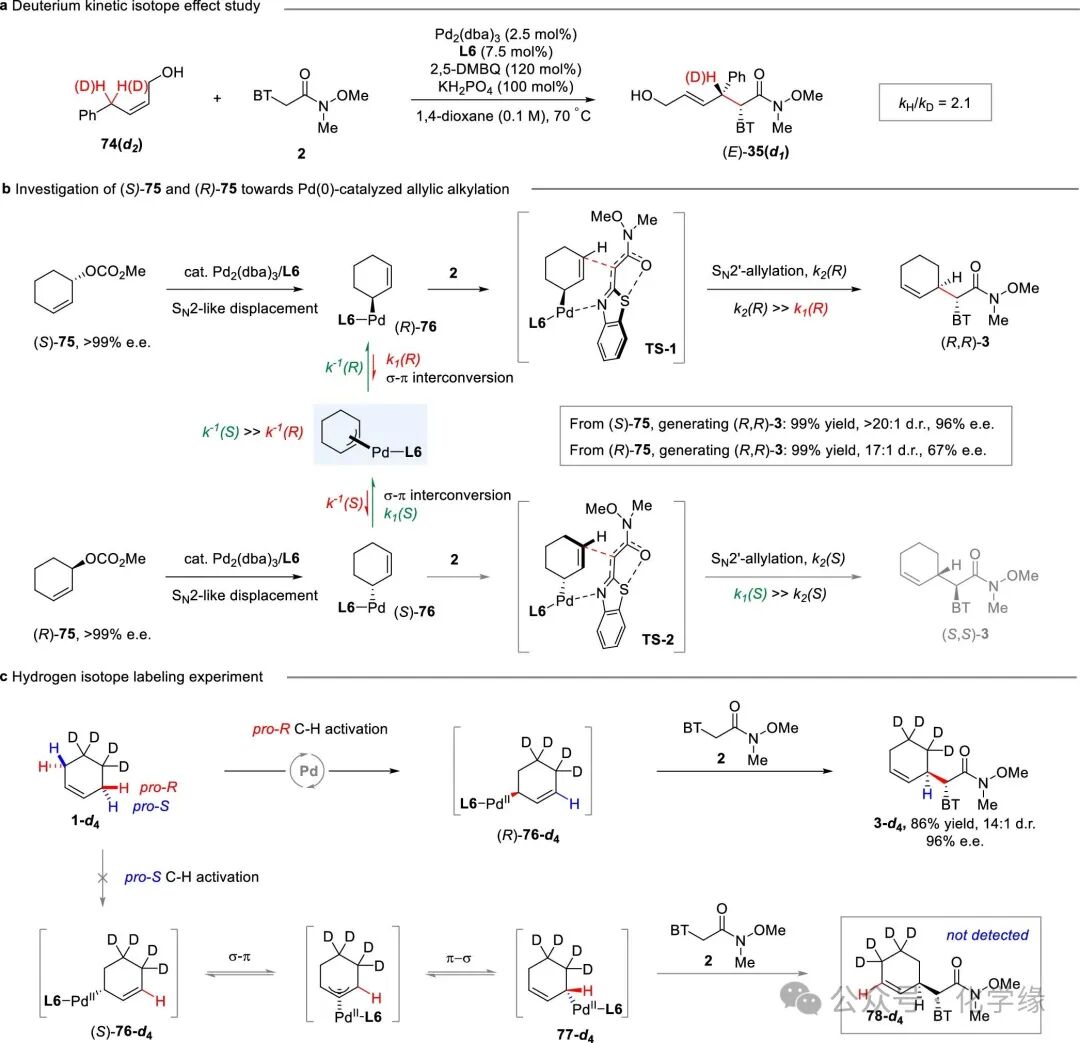
Figure 8 Mechanistic studies
To elucidate the reaction mechanism, a series of kinetic studies and control experiments were conducted. Initially, the kinetic isotope effect (KIE) of the reaction of (Z)-4-phenyl-2-butanol (d2) (74-d2) and α-benzenesulfonamide 2 was observed, yielding a KIE of kH/kD=2.1, indicating that the cleavage of the allylic C-H bond is likely the rate-limiting step.
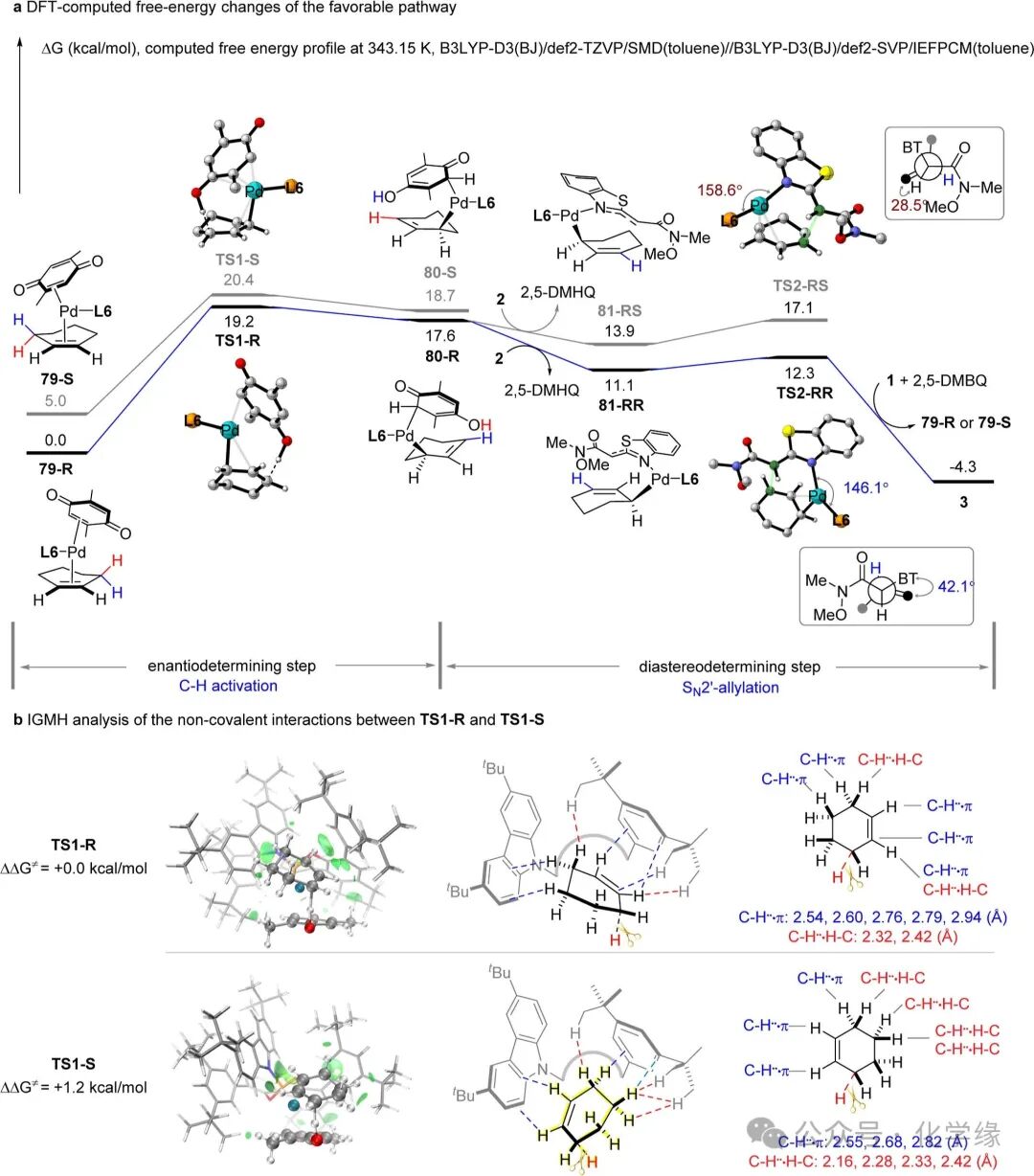
Figure 9 DFT calculations
To gain deeper insights into the origin of stereoselectivity at the molecular level, density functional theory (DFT) calculations were comprehensively conducted on the catalytic cycle. Based on previous studies, it is hypothesized that the catalytic cycle begins with the complexation of cyclohexene 1, 2,5-DMBQ, and the chiral phosphoramidite-Pd catalyst, generating 16-electron Pd(0) species 79-R and 79-S. The energy of 79-R is more favorable, with its stability being 5.0 kcal/mol higher than that of 79-S, and its corresponding transition state for cooperative proton and two-electron transfer TS1-R is lower than TS1-S by 1.2 kcal/mol. This initial step effectively determines the preferential formation of chiral σ-allyl-Pd intermediates 80-R. Subsequently, α-benzenesulfonamide 2 undergoes deprotonation with 80-R, ultimately forming ion pair complexes 81-RR and 81-RS. The formation of C-C bonds proceeds via an inner-sphere SN2′ allylation pathway, determining the non-diastereoselectivity. The pathway leading to the formation of 3 through TS2-RR is more favorable than that through TS2-RS by 4.8 kcal/mol, consistent with the observed excellent enantioselectivity. Comparing TS2-RR and TS2-RS, the palladium-nucleophile complex in TS2-RS undergoes significant distortion to form the C-C bond, thus favoring the adoption of TS2-RR (P-Pd-O bond angle: TS2-RS is 158.6°, while TS2-RR is 146.1°). This distortion is also evident in Newman projection analysis, where the dihedral angle of transition state TS2-RS (28.5°) significantly deviates from the stable staggered conformation, while TS2-RR (42.1°) is distinctly different. A comprehensive analysis of this process indicates that the key step determining the generation of enantiomers and limiting the reaction rate is the activation of the allylic C-H bond through the intermediate TS1-R, with a total energy barrier of 19.2 kcal/mol, consistent with experimental kinetic studies. Furthermore, the methyl group in TS2-RR forms two stabilizing C–H···H–C interactions with the ligand pocket, collectively imparting unique efficacy to the Weinreb amide as a nucleophile.
Conclusion: This study successfully demonstrates an efficient method for site- and enantioselective allylic C-H alkylation of internal alkenes using pocket-like chiral phosphoramidite-Pd catalysis. The broad applicability of this method has been validated through the functionalization of various cyclic and acyclic alkenes, and it is suitable for a range of alkenes conjugated with pharmacologically important molecules. The key to this method lies in the introduction of an enzyme-like chiral environment through the use of pocket-like chiral phosphoramidite ligands, which stabilize the transition states that determine enantiomeric outcomes, thereby facilitating the selective activation of similar allylic C-H bonds. Mechanistic studies indicate that the rapid SN2′-allylation pathway surpasses the σ-π tautomerization process, ensuring that functional groups are precisely installed at the initially cleaved prochiral hydrogen positions. This method, achieved through pocket-like chiral phosphoramidite-Pd catalytic systems, opens new avenues for further exploration of site-selective chiral allylic C-H functionalization reactions.
Article Information:
Palladium-Catalyzed Site– and Enantiodifferentiating Allylic C–H Alkylation of Internal Alkenes
Zhi-Peng Shi, Zi-Han Lin, Ye-Xuan Xu, Long-Fei Fan, Pu-Sheng Wang*, Xin Hong*, Liu-Zhu Gong*
DOI: 10.1021/jacs.5c13527
We hope this article inspires you, and let us work together for a better future!
