
Nonaqueous lithium-oxygen batteries (LOBs) have significant application potential due to their extremely high theoretical energy density, but the role of the electrolyte in regulating the reaction kinetics during the oxygen reduction reaction (ORR) remains unclear. Here, we systematically investigate the ORR pathways at the electrolyte interface with Co-N-C single-atom catalysts (SACs) using ab initio molecular dynamics (AIMD). In the bulk electrolyte, simulation results show that the solvent binding strength of Li+-solvent follows the order of donor number (DN), which may lead to differences in the free energy of interfacial reactions. High DN solvents increase the Li+ insertion barrier due to strong Li+-solvent binding, while promoting the desorption of *LiO2. However, it is found that the desorption of *LiO2 into the electrolyte is thermodynamically unfavorable, thus driving the reaction towards surface-mediated growth. Low DN electrolytes combined with high-adsorption catalysts promote surface growth, while high DN systems combined with weak-adsorption catalysts favor solution growth. Our study proposes a “catalyst-electrolyte interface lithium ion competition” principle that determines the formation pathway of discharge products and provides strategies for the optimized design of lithium metal batteries.
Research Background
Nonaqueous lithium-oxygen batteries are considered strong candidates for next-generation high-energy storage devices due to their ultra-high theoretical energy density (~3500 Wh/kg). However, their practical application is limited by the slow kinetics of the oxygen reduction reaction and oxygen evolution reaction, leading to severe polarization and short cycle life during charge and discharge processes. The electrolyte plays a key role in regulating the ORR reaction pathway and product formation mechanism, but its microscopic action mechanism remains unclear.
Current research on LOBs cathode catalysts mainly focuses on the relationship between the materials themselves (such as Pt, oxides, carbon materials, etc.) and the adsorption energy of intermediates, with most computational models neglecting the electrolyte environment and basing the catalytic mechanism solely on vacuum conditions. Experiments have shown that the donor number of the electrolyte significantly affects the solubility of the intermediate LiO₂ and the morphology of the product Li₂O₂, but there is a lack of atomic-scale mechanistic understanding.
This study combines ab initio molecular dynamics with free energy sampling for the first time to systematically investigate the effect of electrolyte DN values on the ORR pathways at the Co–N–C single-atom catalyst interface, revealing the Li⁺–solvent-catalyst tri-competition mechanism, providing theoretical basis and optimization strategies for rational design of electrolyte-cathode synergistic systems.
Scientific Questions
How does the electrolyte DN value affect the transport of Li⁺, the desorption of *LiO₂, and the growth pathway of Li₂O₂ (surface growth vs. solution growth) during the ORR process?
Research Plan
1. Use VASP for AIMD simulations, employing the PBE-D3 functional, NVT ensemble, at 300 K;
2. Construct three electrolyte models (DMSO/DME/MeCN + LiTFSI + O₂);
3. Use the Blue Moon slow growth method to calculate free energy, with reaction coordinates including Li–O and Co–O distances;
4. Analyze solvation structure (RDF, coordination number), reaction energy barriers (Eₐ), and free energy changes (ΔF).
Innovations
1. Methodological Innovation: For the first time, AIMD is combined with free energy sampling for the study of the ORR at the solid-liquid interface of LOBs;
2. Mechanistic Innovation: Propose the “Li⁺ competition mechanism”, clarifying how the DN value influences the growth pathway by modulating the interactions between Li⁺-solvent and Li⁺-catalyst;
3. Strategic Innovation: Propose optimizing battery performance by adjusting the catalyst adsorption energy to match the electrolyte DN value.
Figure 1
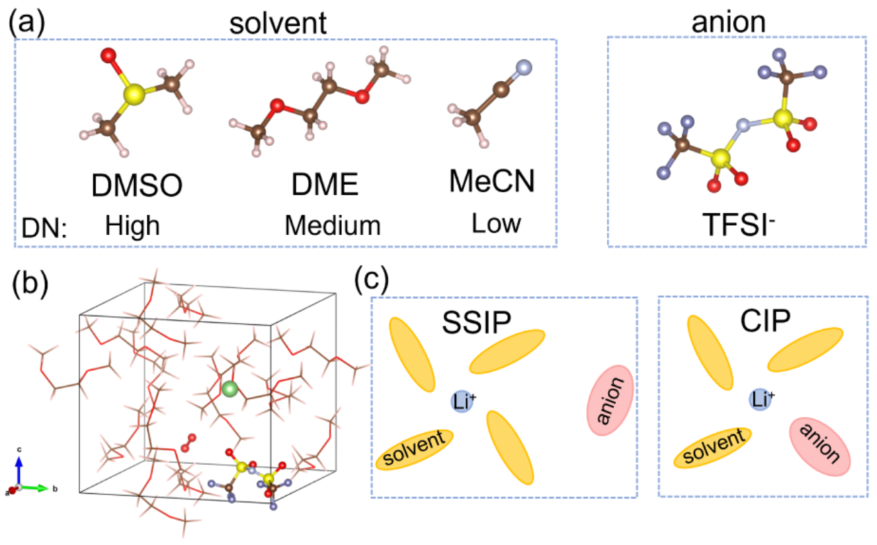
1a: Shows the molecular structures of the TFSI⁻ anion and three solvents (DMSO, DME, MeCN).
1b: Displays the structure of the electrolyte simulation box based on DME, containing Li⁺, TFSI⁻, O₂, and solvent molecules, reflecting the actual electrolyte environment.
1c: Schematic diagram distinguishing between SSIP (solvent-separated ion pairs) and CIP (contact ion pairs) of the initial coordination mode of Li⁺.
Figure 2
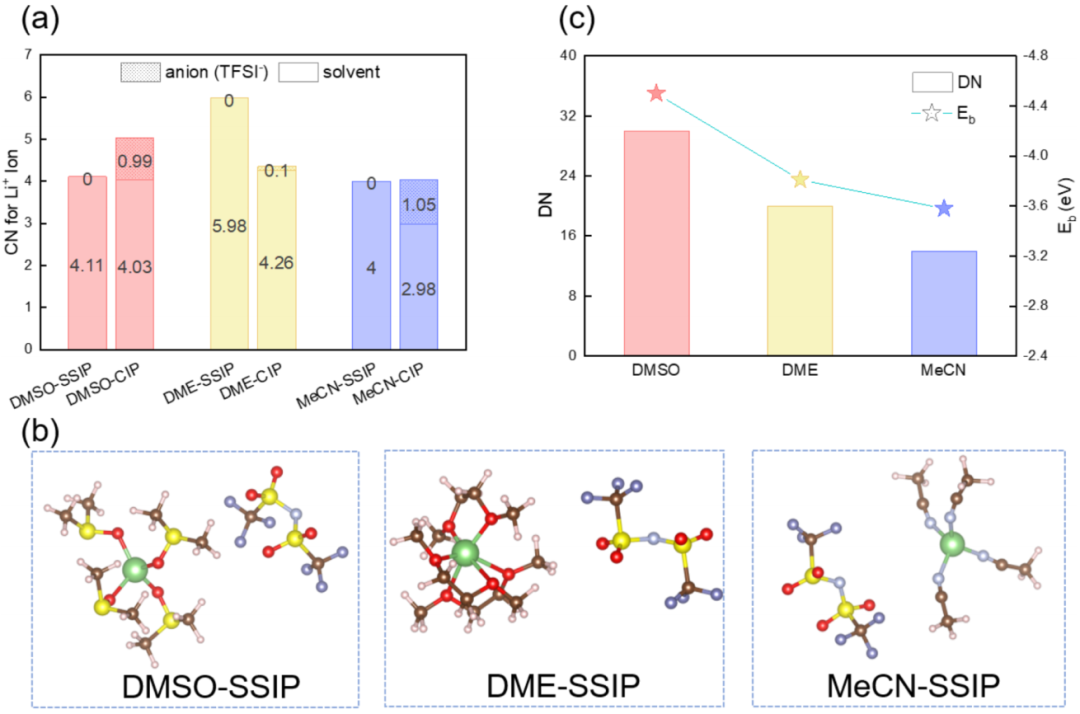
2a: Shows the coordination of Li⁺ in the first coordination layer under the initial configuration of SSIP, with DMSO having 4.11 O, DME having 5.98 O, and MeCN having 4 N.
2b: Shows Li⁺ in DME forming a loose spherical structure with three bidentate coordination, with a coordination radius of 3.75 Å.
2c: The solvent binding energy (Eₑ) of Li⁺ is positively correlated with the DN value, with DMSO being the strongest (-4.50 eV), and MeCN being the weakest (-3.58 eV).
Figure 3
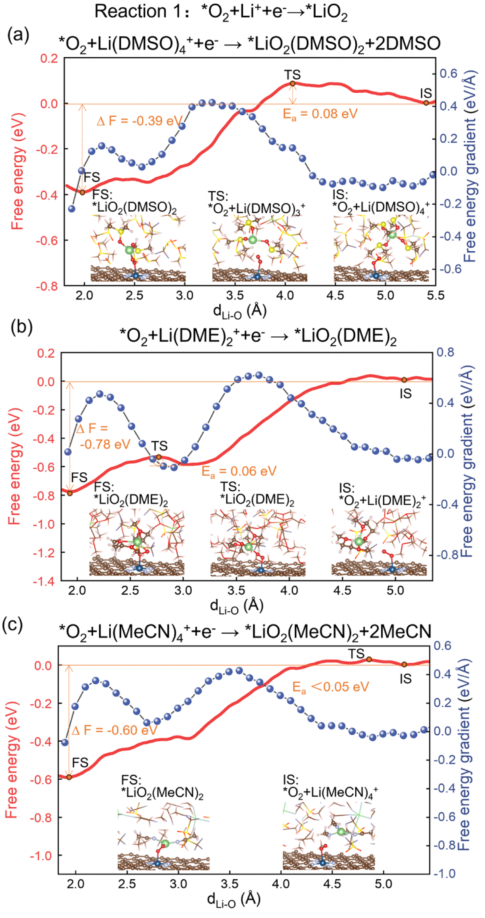
ShowsReaction 1 (*O₂ + Li⁺ + e⁻→ *LiO₂) free energy profile:
3a (DMSO): Eₐ = 0.08 eV, ΔF = –0.39 eV;
3b (DME): Eₐ = 0.06 eV, ΔF = –0.78 eV;
3c (MeCN): Eₐ < 0.05 eV, ΔF = –0.60 eV.
This indicates that low DN electrolytes favor the transfer of Li⁺ to *O₂.
Figure 4
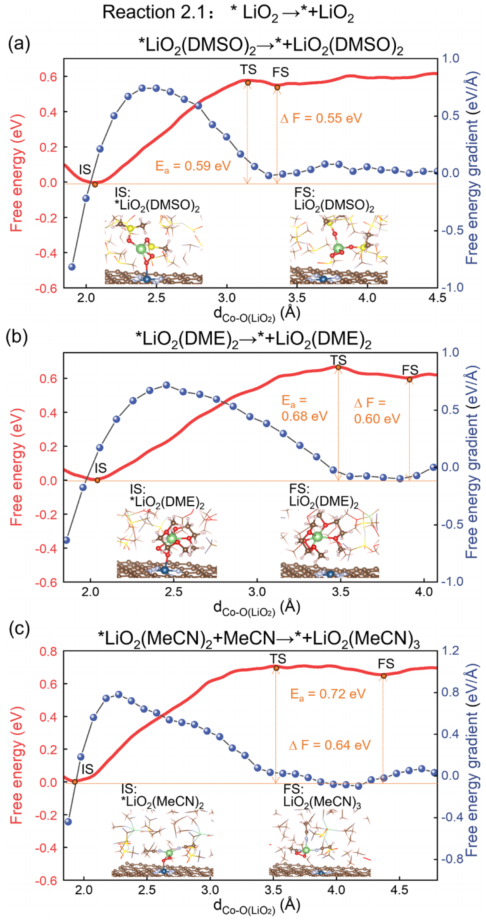
ShowsReaction 2.1 (*LiO₂→ LiO₂(sol)) free energy profile:
4a (DMSO): Eₐ = 0.59 eV, ΔF = +0.55 eV;
4b (DME): Eₐ = 0.68 eV, ΔF = +0.60 eV;
4c (MeCN): Eₐ = 0.72 eV, ΔF = +0.64 eV.
*LiO₂ desorption is thermodynamically unfavorable in all systems, with high DN slightly reducing the energy barrier.
Figure 5
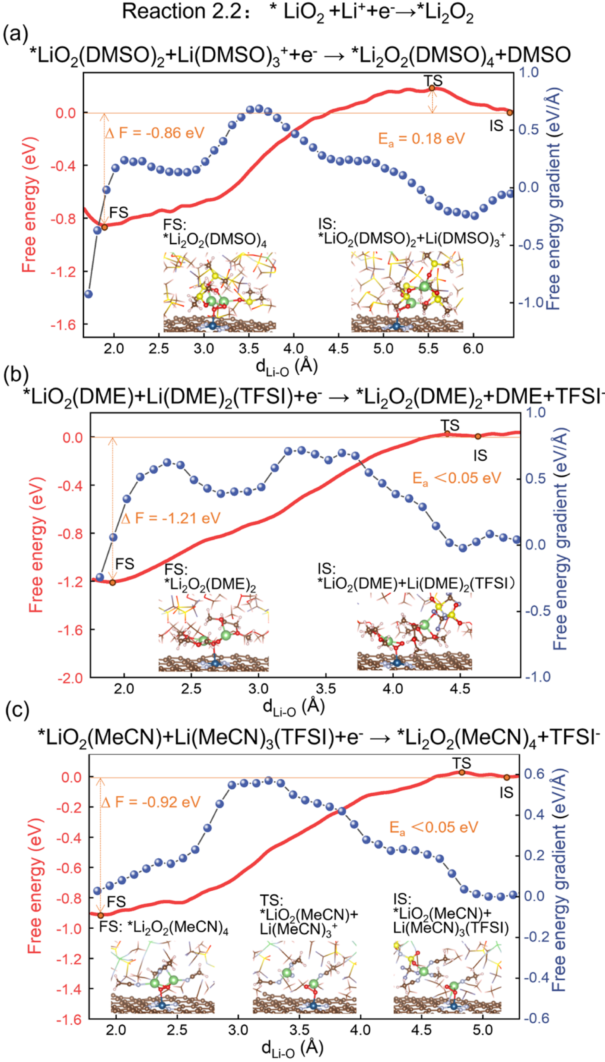
ShowsReaction 2.2 (*LiO₂ + Li⁺ + e⁻→ *Li₂O₂) free energy profile:
5a (DMSO): Eₐ = 0.18 eV, ΔF = –0.86 eV;
5b (DME): Eₐ < 0.05 eV, ΔF = –1.21 eV;
5c (MeCN): Eₐ < 0.05 eV, ΔF = –0.92 eV.
This indicates that the surface growth pathway is favored both thermodynamically and kinetically.
Figure 6
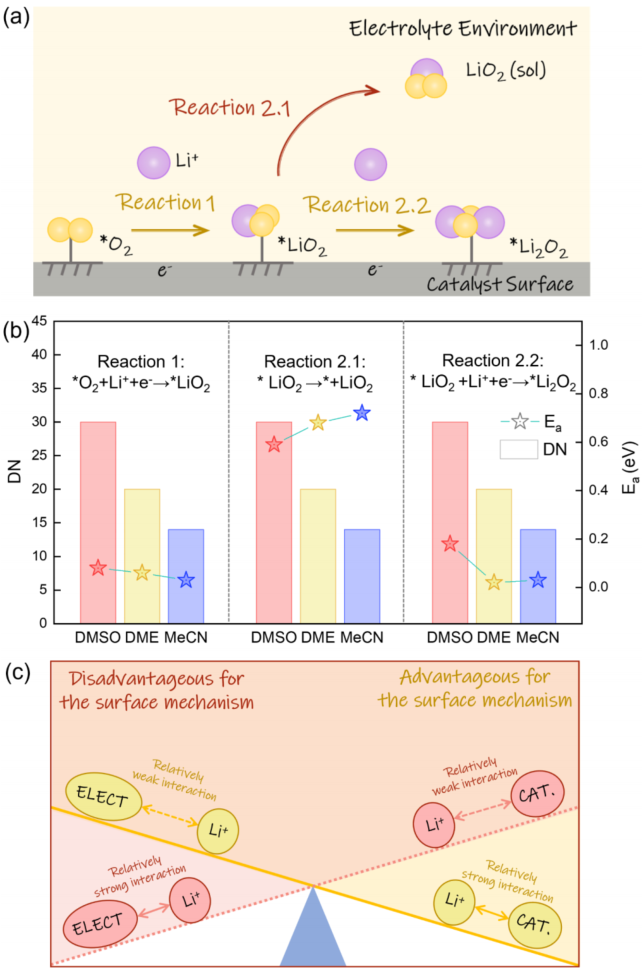
6a: Schematic diagram of three key pathways for the ORR (Li⁺ addition, LiO₂ desorption, Li₂O₂ formation).
6b: Graph showing the relationship between Eₐ and ΔF with DN values, indicating that high DN increases the Li⁺ addition barrier and reduces the *LiO₂ desorption barrier.
6c: Schematic diagram illustrating how the competition between Li⁺-solvent and Li⁺-catalyst interactions determines the growth mechanism.
Core Conclusions
1. Li⁺ exists mainly in the form of SSIP in all electrolytes, with high DN enhancing the Li⁺-solvent binding but increasing the interfacial Li⁺ transport barrier;
2. *LiO₂ desorption is thermodynamically unfavorable in all systems (ΔF > 0.55 eV), with the surface growth pathway dominating;
3. Low DN electrolytes (such as MeCN) promote surface growth, while high DN electrolytes (such as DMSO) slightly favor solution growth but are limited by the *LiO₂ adsorption strength;
4. It is proposed that optimizing the growth pathway of Li₂O₂ and battery performance can be achieved by adjusting the catalyst adsorption energy to match the electrolyte DN value.
Paper Information:
Unveiling Electrolyte Effects on the Oxygen Reduction at Co−N−C Single-Atom Catalysts in Nonaqueous Lithium−Oxygen Batteries by Ab Initio Molecular Dynamics
https://doi.org/10.1021/acscatal.5c03920
Disclaimer: This post is for academic sharing only and does not involve any commercial activities. If there are any copyright issues or inappropriate descriptions, please contact the editor to delete.