When writing the previous article “TCE, Structure and Function, Bioassay Design,” I remained firmly in the “silent Fc” camp regarding TCE molecules. I wrote:
For the new TCE molecules, it is generally preferred to choose a structural design with a silent Fc segment. By incorporating the Fc segment design, one can gain the advantage of a long half-life, while the silent Fc segment design can minimize non-specific immune activation mediated by Fc receptors.
I also believe that a TCE molecule does not need the Fc to exert any effects other than FcRn binding. This is determined by the mechanism of action of TCE. The tumor cell-killing ability of TCE is entirely derived from the recruited T cells, rather than the functions of other immune cells (such as ADCC, ADCP, CDC). On one hand, we do not want to cause damage to cells other than tumor cells (especially the recruited T cells), and on the other hand, we want to avoid the risks of uncontrolled FcγR-mediated crosslinking and non-specific immune activation. With Fc silenced, there is no need to consider other killing pathways.
So when I saw the design of TCE-ADC this year, my brain was overloaded for a moment. TCE? Can it also be made into an ADC?
Can DLL3×CD3 TCE also be made into an ADC?
This design comes from Leads Biolabs’ LBL-058, which is formed by coupling a DLL3-targeting TCE with a TOP1 inhibitor (TOP1i) payload. They call it a T cell engager conjugate (TEC). Undoubtedly, this is first-in-class.
Without discussing the idea of directly making TCE into an ADC, it is not uncommon for TCE and ADC to be used in combination in clinical settings. For example, Roche’s CD20×CD3 TCE (mosunetuzumab) is used in combination with CD79b ADC (polatuzumab vedotin) to treat relapsed or refractory large B-cell lymphoma, Merck’s DLL3×albumin×CD3 TCE (MK-6070) is used in combination with Daiichi Sankyo’s B7H3-ADC (DS-7300) to treat small cell lung cancer (SCLC), and Amgen’s DLL3×CD3 TCE (Tarlatamab) is used in combination with Yiling Biotech’s B7-H3 ADC (YL201) to treat SCLC. Focusing on the field of small cell lung cancer (SCLC), DLL3 is undoubtedly a shining target. The TCE drug, the DLL3×CD3 TCE Tarlatamab, developed by BeiGene and Amgen, was approved by the FDA last May, and its marketing application in China was accepted this year, being included in the priority review process. The ADC drug, ZL-1310 from Zai Lab, received FDA fast track designation this May, and Roche’s DLL3 ADC in collaboration with Innovent Biologics is also planned to initiate global Phase III clinical trials in 2026.
Why design multiple drugs with different mechanisms of action for the same target? The commercial landscape is still uncertain, so here I will share my understanding from the perspective of mechanisms of action.
Let’s take TCE as an example (using a somewhat informal artistic description). During TCE therapy, one end of the TCE molecule binds to the surface of the tumor cell, while the other end recruits T cells for killing. In response, tumor cells upregulate the expression of immunosuppressive ligands such as PD-L1 to evade this killing, inhibiting T cell activity through the binding of PD-L1 to PD-1 on T cells. This is one way tumors develop resistance. If we want to improve this resistance, we can consider combination therapy with anti-PD-1 or anti-PD-L1 drugs to relieve the tumor cell’s suppression of T cells. To counteract our suppression of T cells, tumor cells may even downregulate the expression of the targeted tumor antigen, further developing resistance. If we want to improve this resistance, we need to consider targeting other antigens in the tumor or other therapeutic strategies (such as targeting the tumor microenvironment or utilizing bystander effects).
It is easy to understand that if two different therapeutic mechanisms are employed simultaneously, the likelihood of tumor cells evolving resistance to both mechanisms is low, thus combination therapy may yield better efficacy (case by case). We have many options, such as TCEs that recruit T cells for killing, ADCs that kill through endocytosis, immune checkpoint inhibitors that block and relieve immunosuppression, and other antibody drugs like anti-VEGF. How to combine these to achieve the best effect is a question that needs consideration.
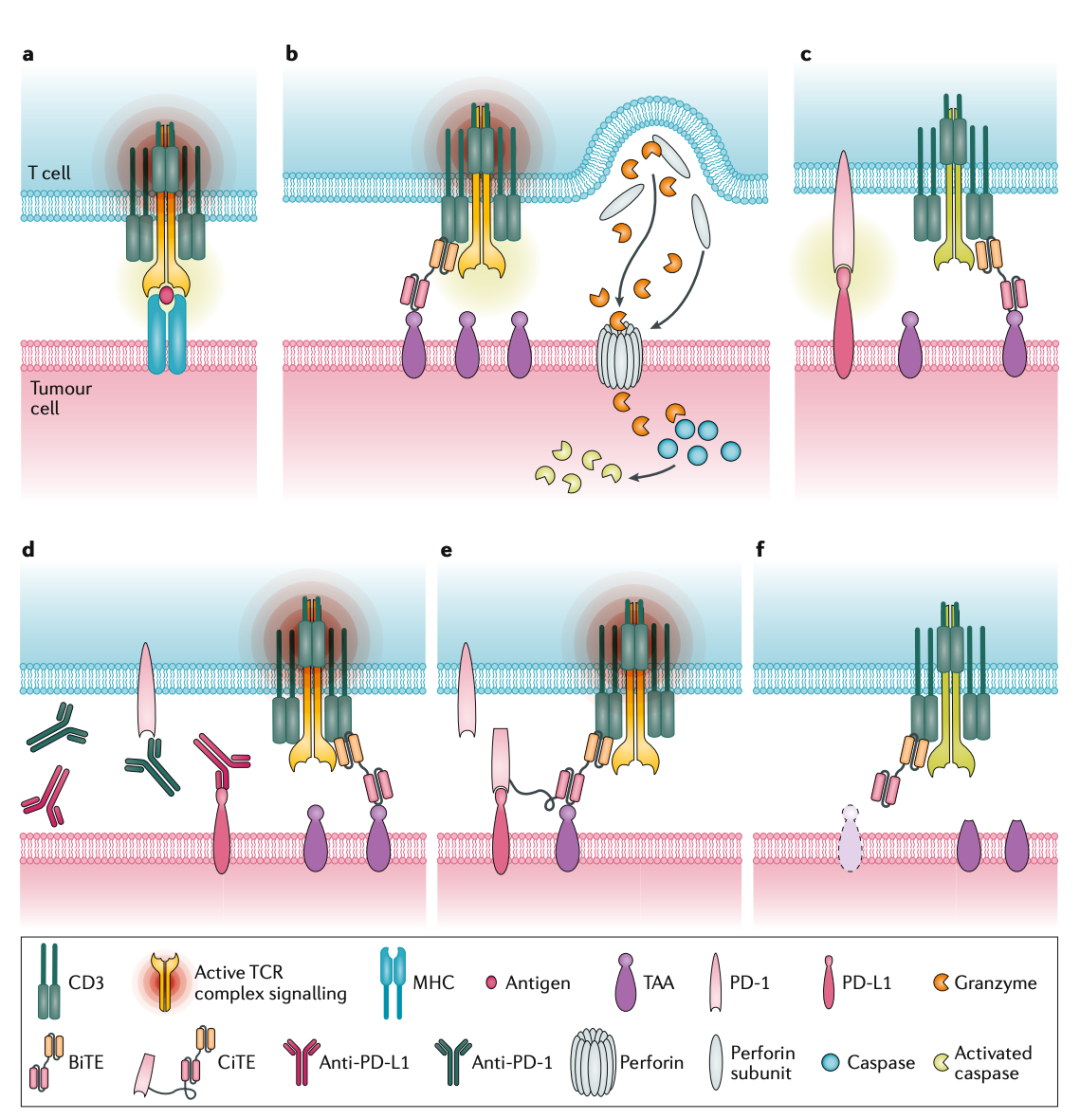
Now, the idea of directly making TCE into an ADC does not seem completely incomprehensible. I believe the design of the DLL3×CD3 ADC aims to simultaneously leverage the therapeutic advantages of two mechanisms to overcome the resistance issues associated with single DLL3-targeted therapies.
Leads’ TEC is a product of the LeadsBody platform and the TOPiKinetics platform. By reducing CD3 affinity and optimizing the spatial structure of the molecule, it enhances the conditional activation of T cells, avoiding some of the issues mentioned earlier with TCE therapies. Additionally, it enhances the affinity for the DLL3 target, enabling rapid endocytosis to exert killing functions and utilizing the TOP1 inhibitor toxin to achieve bystander killing effects, addressing the impact of tumor antigen expression heterogeneity on treatment.
At this point, I also feel that with this complex mechanism of action, in vitro biological activity experiments are becoming troublesome. Let me make some random guesses. First, as is customary, binding activity methods will be established, such as DLL3×CD3 binding ELISA. This typically poses no issues for quality control of TCEs in early stages. Second, it is necessary to establish activity methods for TCE and ADC functions separately, based on cellular TDCC and cytotoxicity methods, which should not be problematic. The TDCC method based on reporter genes is already quite mature, with a short experimental cycle (4-6 hours); while cytotoxicity assays without T cells take longer (~72 hours), and due to the different time windows, the two mechanisms will not significantly interfere with each other in experiments. These two methods can be used in the release and stability throughout the product lifecycle. As for endocytosis efficiency, bystander effects, and other safety experiments, they can be selectively conducted during characterization.
PD-L1 Immune Checkpoint, Let’s Make it an ADC Too
This year, PD-L1 ADC drugs are also very popular. Hualan Biological’s HLX43 and Pfizer’s PF-08046054 (SGN-PDL1V) both appear to have good clinical performance. When I saw that the PD-L1 target was also made into an ADC drug, I was not too surprised. After all, designing a tumor surface highly expressed membrane protein as an ADC target is not an outrageous choice. What concerns me more is how PD-L1, being a broadly expressed target, can avoid killing “good citizens” like activated T cells, APC cells, and normal tissue cells while killing tumor cells and even Treg cells.
What I can think of is to apply the TCE approach mentioned earlier, by reducing the affinity for PD-L1 in the molecular design, or designing two low-affinity PD-L1 binding domains based on the differences in PD-L1 expression levels between “good citizen” cells and tumor cells to enhance affinity for high-expressing PD-L1 cells.
Of course, I am not aware of the affinity design of the PD-L1 ADC currently under investigation. At least in the publicly available data, Hualan Biological’s HLX43 does not show reduced affinity compared to its naked antibody HLX20.
By chance, I also came across an interesting mechanism called MDR1 Efflux Pump. If I were familiar with chemotherapy, I might have learned about this concept earlier. MDR1 is a common and classic mechanism of chemotherapy resistance.MDR1, also known as P-gp (P-glycoprotein), is an efflux pump located on the cell membrane. Its main function is to pump various exogenous or endogenous hydrophobic small molecules out of the cell, thereby protecting the cell from toxic substances. In activated T cells, MDR1 expression is often upregulated. This means that even if activated T cells endocytose the ADC and release the payload (or the payload from bystander effects), they can expel the toxin through the MDR1 efflux pump, thereby reducing killing. Moreover, the expression level of PD-L1 on these cells is still far lower than that on tumor cells, and the endocytosis of ADC is, after all, a minority.
However, MDR1 is not omnipotent; it is only effective against certain small molecule toxins. Microtubule inhibitors (such as MMAE, MMAF, DM1, etc.) and some topoisomerase inhibitors (such as SN-38) are usually substrates of MDR1 and can be pumped out of the cell by MDR1. Some newly designed topoisomerase I inhibitors specifically designed to overcome resistance, such as DXd (a derivative of exatecan) (thanks to the comments for the reminder, DXd is also a substrate of MDR1), have structures optimized to typically not be substrates of MDR1 (which also allows them to show better efficacy in treating tumors resistant to traditional chemotherapy drugs). Therefore, whether to “utilize” or “avoid” the MDR1 mechanism in selecting small molecule toxins is another issue that requires weighing. For example, Pfizer’s PF-08046054 (SGN-PDL1V) uses the classic vc-MMAE as a linker-payload, with a DAR of 4.
In contrast, Hualan Biological’s HLX43 is conjugated with an optimized topoisomerase I (TOP1i) inhibitor C24, with a DAR of about 8. In the linker-payload design, HLX43 is designed to cleave and release toxins in the tumor microenvironment (from Yiling Biotech’s TMALIN® technology platform), and does not need to rely entirely on endocytosis (of course, efficient endocytosis is still the main release pathway). This design can further reduce the impact of ADC on normal cells.
PD-L1 ADC drugs, while also having the effect of PD-L1 blockade, primarily rely on endocytosis and toxin release to exert their effects. The dosing of immune checkpoint antibodies is above 10 mg/kg, significantly higher than the clinical dosing of ADC drugs. Therefore, for ADCs, the blocking effect is not the main MoA, which is very reasonable. Continuing along this line of thought, PD-L1 ADC drugs will also have good synergistic effects with PD-1 antibodies. This has also been confirmed in clinical trials.
As for in vitro biological activity, thanks to the not overly complex mechanism of action, the activity methods will not be too troublesome and can follow the conventional ADC drug approach. Binding activity + cytotoxicity is sufficient for quality control. Endocytosis efficiency and bystander effects, as well as safety for “good citizen” cells, can be used for further in vitro characterization.
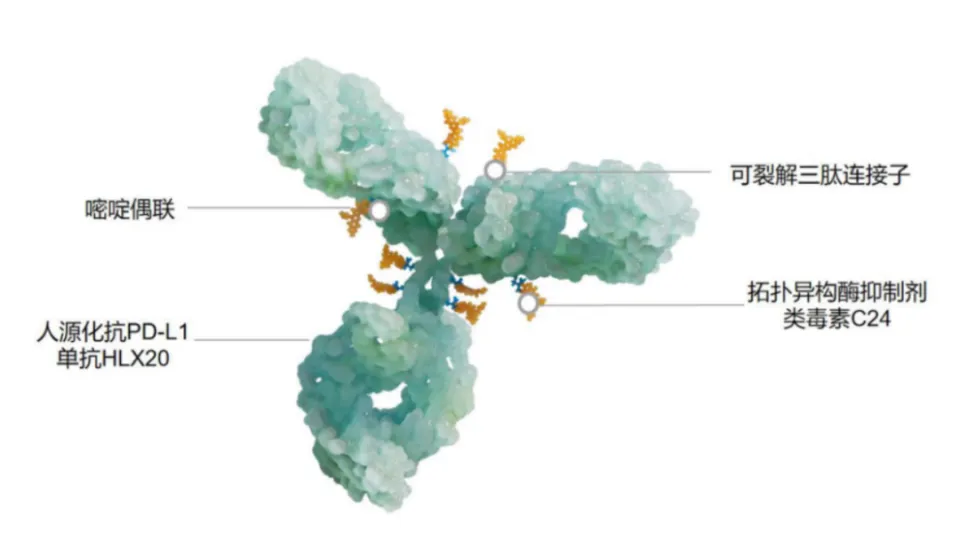
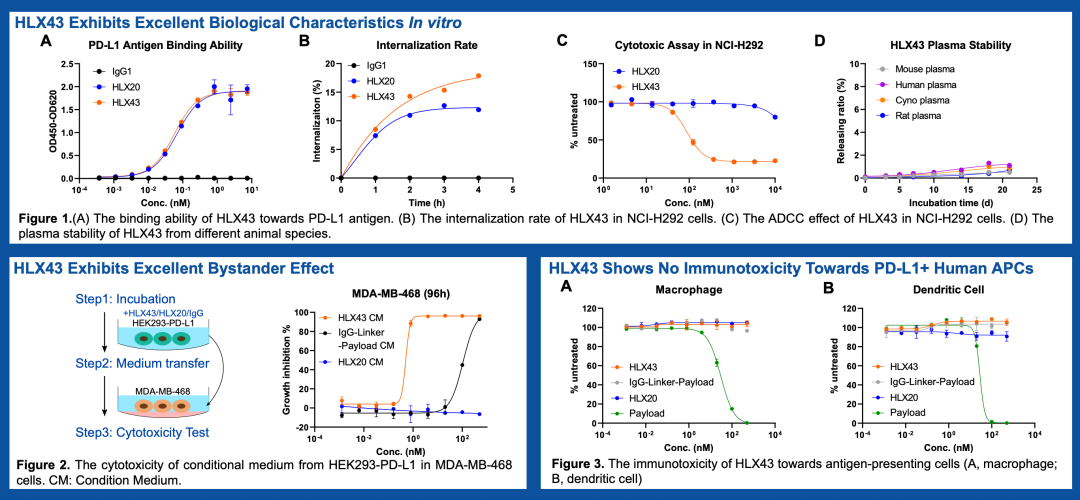
VEGF, Well, It Can Also Be an ADC Target
This year, I have seen many strange ADC targets. VEGF can also be an ADC target.
We all know VEGF, vascular endothelial growth factor. When tumor cells are hypoxic, they secrete large amounts of VEGF to attract and stimulate nearby endothelial cells to proliferate, migrate, and rearrange, ultimately forming new blood vessels to solve the problem of oxygen and nutrient deficiency, ensuring their survival and expansion. We are all familiar with Bevacizumab, which can specifically bind and neutralize VEGF, thereby cutting off the blood and nutrient supply to tumors, starving them and inhibiting their growth and metastasis.
The question is, VEGF is not a membrane protein on the surface of tumor cells, so why can it also be used as an ADC?
This reminds me of when I was working on Bevacizumab biosimilars. At that time, I had to characterize ADCC and CDC effects with limited knowledge, and since VEGF is a free factor rather than a membrane protein, there was logically no suitable target cell to use. Later, I found out that some VEGF can exist in a tethered form on the cell membrane. Following this line of thought, I found some cells with tethered VEGF, and successfully completed the relevant characterization experiments. Of course, tethered VEGF is still a minority, and ADCs will not use this as the main MoA. So what is going on?
VEGF ADC, developed by Yiling Biotech, along with Hualan Biological’s HLX43 mentioned above, is built on Yiling’s TMALIN (Tumor Microenvironment Activable LINker) technology platform. Under this technology platform, the molecules have the ability to undergo extracellular cleavage in the tumor microenvironment, while the toxin-linker design allows the ADC to accumulate in the tumor microenvironment.
This makes it clear that the VEGF ADC is an ADC targeting the tumor microenvironment (TME). Whether it undergoes endocytosis or not is irrelevant; the expression level of the tumor target does not matter; the drug can release toxins in the tumor microenvironment and exert its effects through bystander effects.
This idea is not outrageous; AbbVie previously had a design for a TNF-α ADC (though it did not succeed), but it was coupled with a glucocorticoid receptor modulator, not a small molecule toxin.
However, the in vitro biological experiments, especially the main killing experiment design, become a bit troublesome. For the binding activity of the target protein, this experiment is not difficult to perform. For the killing experiment design, it still needs to focus on the main mechanism of action. If the main mechanism of action is not endocytosis but rather release and bystander effects in the tumor microenvironment, it may be necessary to consider simulating the linker cleavage mechanism of the tumor microenvironment in cell experiments, such as adding specific tissue proteases or adjusting the culture medium pH (depending on the specific mechanism) to conduct cytotoxicity experiments. If endocytosis is also a main mechanism of action, tethered VEGF cells can be used for cytotoxicity experiments. As for the relevant activity characterization, it is relatively conventional.
Conclusion
As the mechanisms of action become increasingly complex, the design of in vitro biological activity also becomes more complicated, even leading to a sense of helplessness. For activity methods, it is crucial that they reflect the MoA, and method performance is equally important. If one method can reflect the MoA but has poor performance, while another method has good performance but limited ability to reflect the MoA, how do we choose? When we have options, we cannot choose a poorly performing method for routine quality control. The pain this brings is multifaceted. Methods that can reflect the MoA, before being sufficiently optimized, can only be used in less demanding characterizations. Sufficient optimization is sometimes limited by the principles of the methods, and sometimes requires waiting for new technologies to mature. Just like Bevacizumab, using Huvec cells for activity method development and use was a disaster. However, after the reporter gene method matured, the activity methods became much easier to use.
Conclusion 2
Perhaps it is due to age, but I have recently fallen into a kind of “obsession,” or this “obsession” suddenly grew from within my body one day. My thinking pattern has become, “Think about why this is happening?” I know that “what has always been done is not necessarily right.” Therefore, I need to define a goal for every action and find a logic for every cause and effect.
But this is exhausting. I try to go with the flow, but find that there are meaningless rules everywhere.
I enjoy using AI tools, from the early Kimi and DeepSeek to now Doubao, ChatGPT, and Gemini (I think ChatGPT Plus is better than Gemini Pro). Until now, aside from some trivial content, I still cannot directly trust the conclusions given by AI on important content; I require it to help me establish the entire logical process to verify its conclusions or to arrive at my own conclusions. I do not like “default premises”; I prefer logically coherent reasoning.