
Author|Wenxin
Small molecule cytotoxins serve as the “warheads” for ADCs and are crucial components in killing tumor cells. Although there are numerous toxins derived from natural products and chemical synthesis, only a small fraction can be used as cytotoxins in ADCs. The reason is that ADCs have high requirements for toxins, which typically must meet several criteria:
(1)The cytotoxicity of the toxin must be extremely high, often requiring effective concentrations at the picomolar or nanomolar level. Studies have shown that after ADCs enter the human body, due to the antibody’s tumor penetration ability, the number of antigen expressions, internalization efficiency, and linker issues, the final dose that reaches each gram of tumor tissue is only 0.0003% to 0.08%. Therefore, highly effective toxins that can kill tumor cells at extremely low concentrations are needed;
(2)The mechanism of action of the cytotoxic agent must be clear. Currently, most ADC design strategies involve releasing the toxin after internalization, so the target of the toxin molecule is best located inside the cell;
(3)The molecular weight of the cytotoxic agent must be relatively small to reduce immunogenicity. Additionally, appropriate solubility is required to facilitate the coupling reaction with antibodies in the buffer solution. Finally, considering the long half-life of ADCs in the circulatory system, the cytotoxic agent should have sufficient stability in plasma.
Early ADCs primarily used conventional chemotherapy drugs as cytotoxins, such as methotrexate, doxorubicin, vincristine, paclitaxel, and their derivatives. However, these ADCs exhibited low activity in clinical settings and did not demonstrate the expected anti-tumor effects, leading to failure. Nowadays, some toxins that are 100 to 1000 times more effective than traditional chemotherapy drugs are being discovered, significantly improving the therapeutic effects of ADCs. Currently, the cytotoxins used in ADCs can be divided into two major categories based on their mechanisms of action: microtubule inhibitors and DNA-damaging agents. Below is a detailed introduction to these categories.
01 Microtubule Inhibitors
1.1 Auristatin Compounds
In the mid-1960s to late 1980s, Professor Pettit’s research group isolated a series of anti-tumor compound families from the Indian Ocean sea hare (Dolabella auricularia), known as auristatin 1 to 15.
Among them, auristatin 10 was the most effective compound at the time, exhibiting the highest anti-proliferative activity against human cancer cell lines (see figure). Through in-depth research, it was found that the mechanism of action of these auristatins involves strong binding to tubulin, inhibiting the formation and polymerization of microtubules, while also blocking GTP hydrolysis, leading to mitotic arrest and ultimately apoptosis of the cells.
In the 1990s, multiple clinical trials for auristatin 10 began. However, during the progression to Phase II clinical trials, severe side effects such as neutropenia and 40% of patients experiencing moderate peripheral neuropathy caused it to withdraw from clinical research. Nevertheless, due to its high potency and therapeutic index in preclinical models, scientists used it as a lead compound to develop water-soluble analogs of auristatin, known as auristatins.
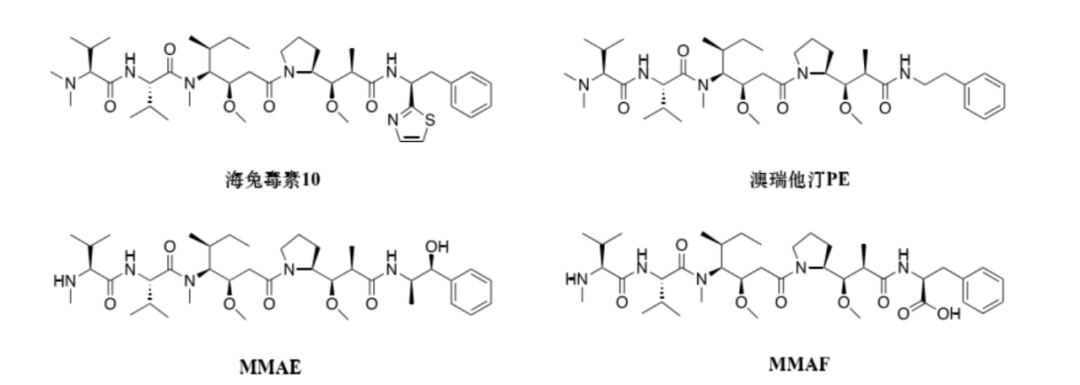 Figure: Structure of Auristatin Cytotoxins
Figure: Structure of Auristatin Cytotoxins
1.1.1 Auristatin PE
Auristatin PE is the first synthetic analog of auristatin 10. Its structural difference is the removal of the thiazole ring from the molecule of auristatin 10 (see figure). Auristatin PE also entered Phase II clinical studies but failed to confirm efficacy in subjects with advanced non-small cell lung cancer and metastatic soft tissue sarcoma, thus no subsequent clinical reports followed.
1.1.2 New Auristatin Derivatives, namelyMMAE and MMAF
In the process of further improving the drug’s in vivo efficacy, two new auristatin derivatives, MMAE and MMAF (see figure), were developed. So far, most ADCs containing auristatin derivatives use MMAE and MMAF as cytotoxins.
Due to the structural differences at the C-terminus of these two derivatives, they possess different physicochemical properties. The high-resolution co-crystal structure of MMAF with tubulin indicates that the carboxyl group at the C-terminus of MMAF can form more hydrogen bonds with amino acids on tubulin, thus giving MMAF superior binding affinity to tubulin compared to MMAE. However, the C-terminus phenylalanine structure composed of the carboxyl group makes MMAF membrane impermeable, thus its in vitro inhibitory activity against cancer cells is about 100 times weaker than MMAE. Furthermore, ADCs using MMAF as the toxin often fail to exhibit the bystander effect.
The N-terminus of MMAF, when linked to an indissoluble linker, forms a linker-toxin complex that often exhibits cytotoxicity similar to that of MMAF, thus it is often used as a toxin in indissoluble ADCs.
Additionally, ADCs using MMAF as the toxin demonstrate inhibitory activity in cell lines with multiple drug resistance (MDR). This indicates that, unlike auristatin 10 and other auristatin derivatives with hydrophobic groups at the C-terminus, MMAF is not a substrate for P-glycoprotein, reducing the sensitivity of the cellular efflux pump. Therefore, ADCs using MMAF as the toxin may have special application value for targeting tumors known to have MDR phenotypes.
Currently, the most representative ADC using auristatin toxins is the marketed Brentuximab Vedotin (brand name, Adcetris), and over half of the ADCs currently in clinical research use this type of toxin.
Some promising ADCs are in clinical trials, including Enfortumab Vedotin (EV-201), Tisotumab Vedotin (GEN701), Ladiratuzumab Vedotin (SGN-LIV1A), Denintuzumab Mafodotin (SGN-CD19A), Polatuzumab Vedotin (DCDS4501A, RG7596), Glembatumumab Vedotin (CDX-011), Depatuxizumab Mafodotin (ABT-414), anti-PSMA ADC, and AGS-16C3F, etc.
1.2 Maytansine Compounds
Maytansine is a 19-membered macrolide structure isolated by Kupchan and his colleagues from the bark of the Ethiopian shrub Maytenus ovatus in 1972 (see figure). Structurally, it is similar to cytotoxins like geldanamycin, rifamycin, and dibromodulcitol. Maytansine can produce significant anti-proliferative effects on most cancer cell lines at sub-nanomolar concentration levels, with an ED50 value ranging from 10-5 to 10-4 μg·mL-1, exhibiting approximately 1000 times higher inhibitory activity than chemotherapeutic drugs like doxorubicin. This makes maytansine a very promising clinical candidate and has been attempted in multiple clinical trials related to cancer treatment.
Unfortunately, due to its lack of specificity, maytansine causes various side effects in patients, such as nausea, vomiting, diarrhea, and neurotoxicity. The low therapeutic index caused by high toxicity means it did not show significant therapeutic effects at tolerable doses, ultimately leading to its withdrawal from clinical trials. Only after the concept of ADCs emerged in the early 1980s, given maytansine’s high toxicity to tumor cells, it was reintroduced into the development and research process as a potential ADC toxin.
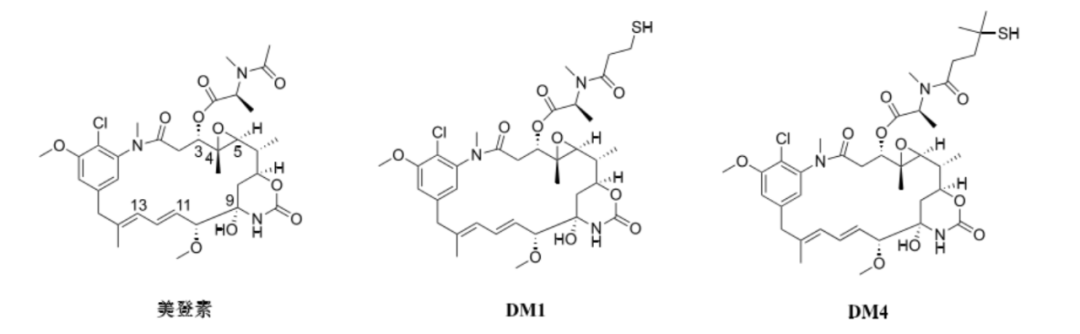 Figure: Structure of Maytansine Cytotoxins
Figure: Structure of Maytansine Cytotoxins
Since maytansine lacks functional groups for coupling with antibodies, some structural modifications are necessary to facilitate its connection with antibodies. Structure-activity relationship studies on maytansine indicate that the hydroxyl and amide groups at C-9, as well as the double bonds at C-11 and C-13, are essential functional groups for activity and cannot be structurally modified. Additionally, the disappearance of the epoxide part will also lead to a decrease in its activity. Therefore, modifications can only be made to the ester side chain at C-3 to produce various maytansine derivatives.
DM1 and DM4 are thiol derivatives modified at C-3 of maytansine and are currently commonly used small molecule cytotoxins in ADCs (see figure). Maytansine and its derivatives are potent microtubule assembly inhibitors, and their mechanism of cytotoxicity is similar to that of vincristine, binding to the same or similar binding sites on tubulin, causing cells to be arrested in the G2/M phase, ultimately inducing apoptosis.
Currently, the FDA-approved cytotoxin Trastuzumab Emtansine (brand name, Kadcyla) is DM1, which has become a leader among maytansine-based ADCs, laying the foundation for the development of more new maytansine-based ADCs targeting different indications. Additionally, several related ADCs are also in clinical research stages, such as Mirvetuximab Soravtansine (IMGN853) for treating FOLR1+ ovarian cancer.
02 DNA-Damaging Agents
2.1 Camptothecin Compounds
Camptothecin is a pentacyclic plant alkaloid isolated from the Chinese native plant Camptotheca acuminata (see figure). Camptothecin and its derivatives can selectively act on DNA topoisomerase I (Topo I), stabilizing the complex formed between DNA and Topo I, preventing broken DNA strands from rejoining, blocking DNA replication and transcription, ultimately leading to apoptosis of tumor cells. However, due to the poor water solubility of camptothecin and the tendency of the lactone ring to open, its bioavailability in vivo is reduced, limiting its clinical application.
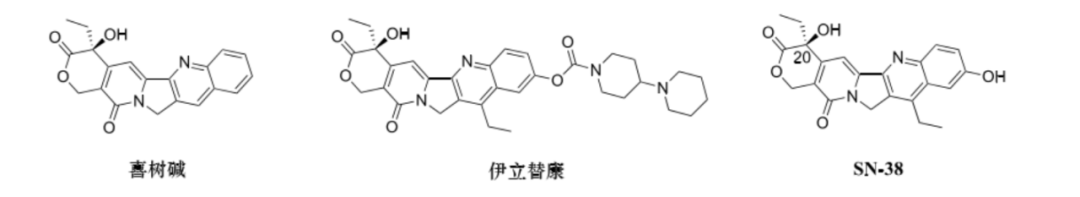 Figure: Structure of Camptothecin Cytotoxins
Figure: Structure of Camptothecin Cytotoxins
To improve the water solubility of camptothecin, water-soluble camptothecin derivatives with piperazine ring prodrugs were developed—irinotecan (see figure). This drug can exist in two forms in vivo: one is in a closed lactone form, and the other is in an open carboxylate form. It is currently approved for the treatment of most metastatic colorectal cancers but also shows efficacy against lung cancer, pancreatic cancer, and breast cancer.
SN-38 is a metabolite of irinotecan and is its main anti-tumor component, with inhibitory activity 2 to 3 orders of magnitude stronger than irinotecan. However, due to its poor solubility and toxicity issues, it can only be administered in prodrug form. The hydroxyl group at C-20 of SN-38 helps reduce the degradation rate of the lactone ring in vivo and can also be linked to the linker for ADC development (see figure).
Labetuzumab Govitecan (IMMU-130) and Sacituzumab Govitecan-hziy (IMMU-132) are both highly therapeutic potential ADCs using SN-38 as the toxin. These two ADCs share the same linker and toxin, with the only difference being the antibodies used, thus they are targeted for different indications. The antibody chosen for IMMU-130 is a humanized anti-CEACAM5 antibody, hMN-14. Currently, it is undergoing Phase II clinical research for the treatment of recurrent or refractory colorectal cancer. The antibody used for IMMU-132 is a humanized antibody hRS7 targeting the Trop-2 antigen, which can be used for the treatment of metastatic triple-negative breast cancer. The overall response rate in Phase II clinical trials for this drug can reach 34%, with 3% of patients achieving complete response (CR). The clinical benefit rate for achieving objective response (OR) and stable disease for more than 6 months is 45%. The median response time, median progression-free survival, and median overall survival (OS) based on statistical calculations show results of 7.6 months, 5.5 months, and 12.7 months, respectively. On February 3, 2023, the U.S. Food and Drug Administration (FDA) approved Sacituzumab Govitecan (SG) for patients with hormone receptor (HR) positive, HER2 negative (IHC 0, 1+ or 2+/ISH -) unresectable locally advanced or metastatic breast cancer who have received prior endocrine therapy and ≥2 lines of systemic treatment (for metastatic disease).
2.2 Calicheamicin Compounds
Calicheamicins are fermentation products obtained from rare actinomycetes in the 1980s by scientists at Lederle Laboratories, which are a class of potent anti-tumor antibiotics acting on DNA. They have been identified as typical enediyne compounds, with complex structures and stereochemistry, composed of a 13-carbon-9-ene-2,6-diyne core, unstable methyl thioether, and an aryl tetrasaccharide chain (see figure).
The aryl tetrasaccharide tail structure makes calicheamicin a highly site-specific DNA cleaver, preferentially embedding into the TCCT-AGGA site of the DNA double helix. Subsequently, the enediyne structure cyclizes via the Bergman reaction, generating active phenyl diradicals that abstract hydrogen atoms from the opposite strand of the DNA deoxyribose backbone, leading to DNA strand breaks. In addition to the TCCT site, other sites such as TTTT, GCCT, TCTC, TCCC, and TCCC can also be cleaved in the same manner.
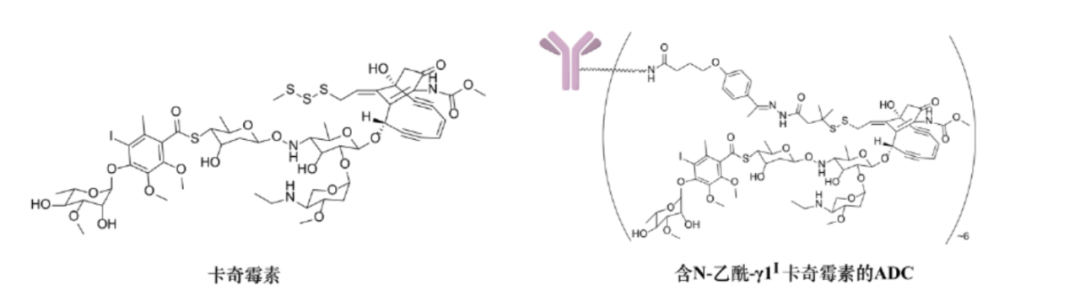
Figure: Structure of Calicheamicin Cytotoxins
Calicheamicin γ1I is currently the most widely studied calicheamicin, exhibiting the strongest cytotoxicity compared to six other similar compounds (α2I, α3I, β1I, β1Br, γ1Br, δ1I) isolated from fermentation broth, being effective at a dose of 0.15 μg·kg-1 against various tumor cells. Although it has shown positive initial experimental results, subsequent evaluations in clinical oncology models revealed a narrow therapeutic window, preventing its clinical application. However, due to its strong cytotoxicity, calicheamicin has become a highly promising cytotoxic agent in the emerging field of ADCs.
In early ADC research, scientists at Lederle Laboratories acetylated the amino sugar structure of calicheamicin γ1I and modified its thioether structure into a disulfide bond to connect with amide or hydrazone linkers, designing a derivative of calicheamicin γ1I, namely N-acetyl-γ1I calicheamicin (see figure). Studies found that this derivative ADC has significant therapeutic advantages compared to the ADC of calicheamicin γ1I, thus it is widely used as a classic toxin in ADCs.
Currently, the FDA-approved Gemtuzumab Ozogamicin (brand name Mylotarg) and Inotuzumab Ozogamicin (brand name Besponsa) both use N-acetyl-γ1I calicheamicin as the toxin.
2.3 PBD Derivatives
In the 1960s, Leimgruber and others isolated an anti-tumor antibiotic called anthramycin from the Streptomyces refuineus, followed by the extraction of more structurally similar molecules with the same function, such as pyrrolobenzodiazepine (PBD) family members from Streptomyces and Micromonospora, such as tomaymycin, DC-81, sibiromycin, and neothramycins.
These analogs share the same structural framework, consisting of fused aromatic rings, diazepine rings, and pyrrole rings (see figure). This structural framework can fit into the grooves of DNA, forming covalent bonds between the electrophilic imine group at N-10/C-11 of the diazepine ring and the amino group at C-2 of guanine in DNA, stabilizing the DNA helix structure, blocking cell division, and thus killing tumor cells.
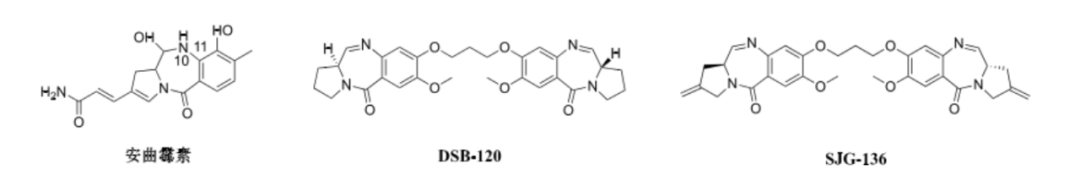
Figure: Structure of Anthramycin Cytotoxins
With the elucidation of the structure-activity relationships of PBD analogs, scientists found that PBD dimers, formed by C-8 connectivity, such as DSB-120 and SJG-136, have a larger interaction surface with DNA, allowing them to form two covalent bonds with guanine, thus fixing DNA more firmly (see figure). This endows PBD dimeric toxins with stronger cytotoxicity, with effective inhibitory concentrations against various tumor cells reaching the picomolar level. Moreover, they also exhibit killing effects on some tumor cells with MDR characteristics.
Since 2013, due to their effective action on DNA, PBDs have gradually been used as new ADC warhead molecules, making them the third emerging toxin after auristatin and maytansine. Currently, more than 10 related ADCs have entered clinical research.
Summary: At this stage, ADC cytotoxins still face the problem of limited variety and single mechanisms of action. Meanwhile, research indicates that the microtubule inhibitors used by most ADCs, such as MMAE and DM1, typically only act on dividing tumor cells and do not significantly kill resting tumor cells, which can lead to reduced therapeutic effects and the emergence of tumor resistance. Therefore, the development of cytotoxins with new mechanisms of action is urgently needed. It is believed that with the continuous efforts of scientists, future small molecule cytotoxins will develop in a more diversified direction, thus providing more powerful and effective “warhead” molecules for ADCs.
03 Overview of Toxins
Through in-depth analysis of over 500 active ADC drug pipelines that have been marketed and are in research, the overview of toxins has been organized. Currently, over 130 types of toxins have been disclosed.
Table 1: Types of Toxins and Pipeline Numbers at Various Clinical Stages
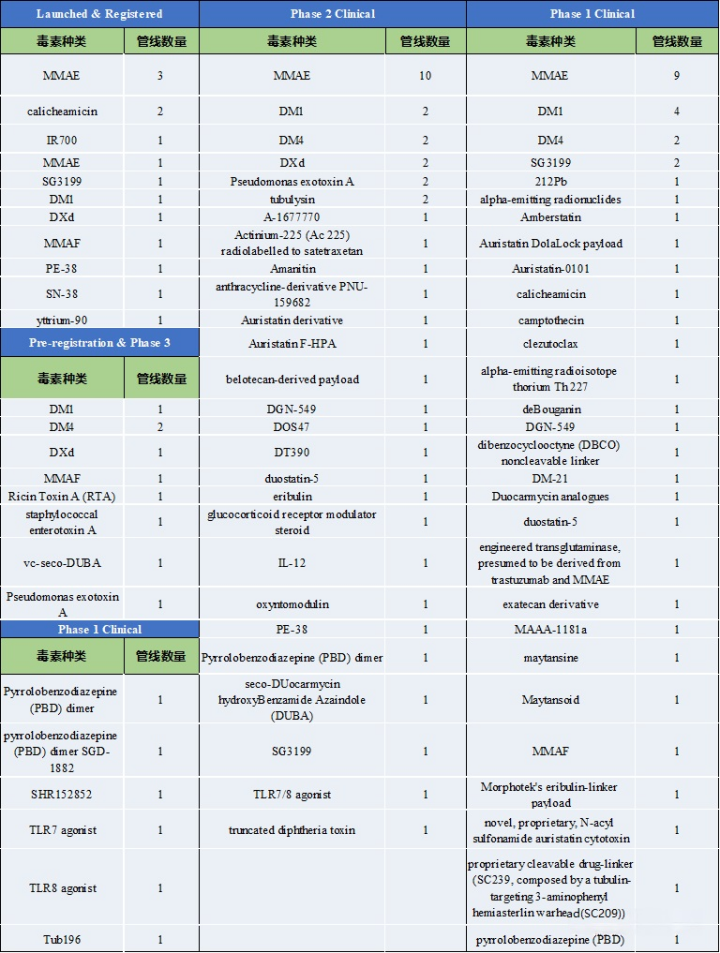
Source: 2022.2 “Overview of ADC Drug Toxins and Linkers”
Table 2: Types of Toxins and Pipeline Numbers at Preclinical Stages
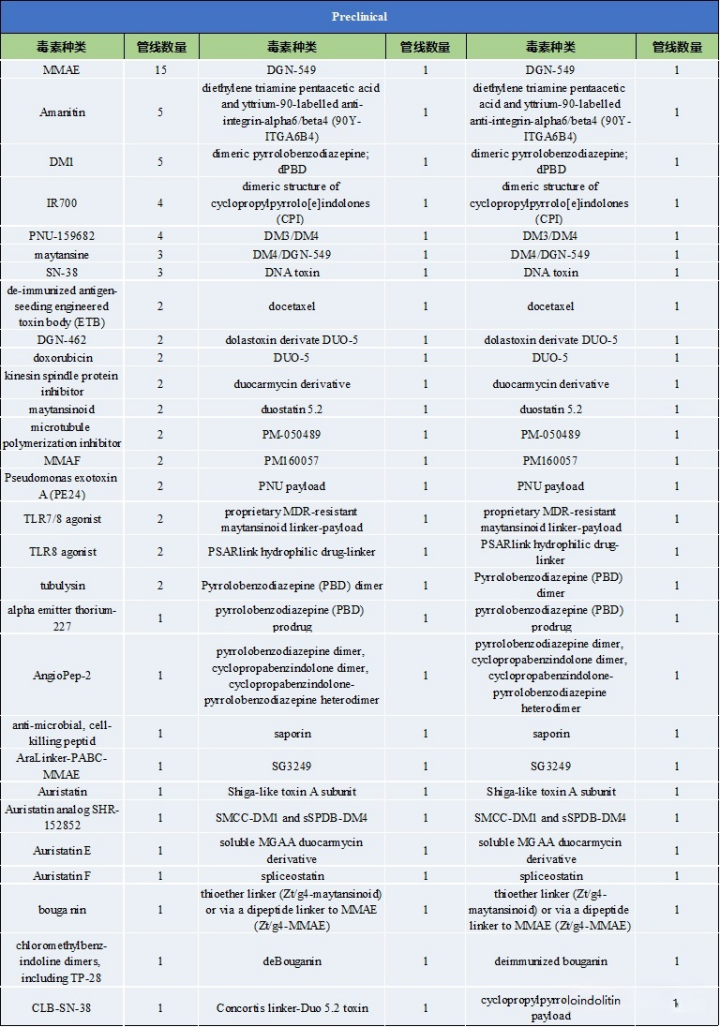
Source: 2022.2 “Overview of ADC Drug Toxins and Linkers”
Table 3: Analysis of Toxins in ADC Drugs at Different Clinical Stages
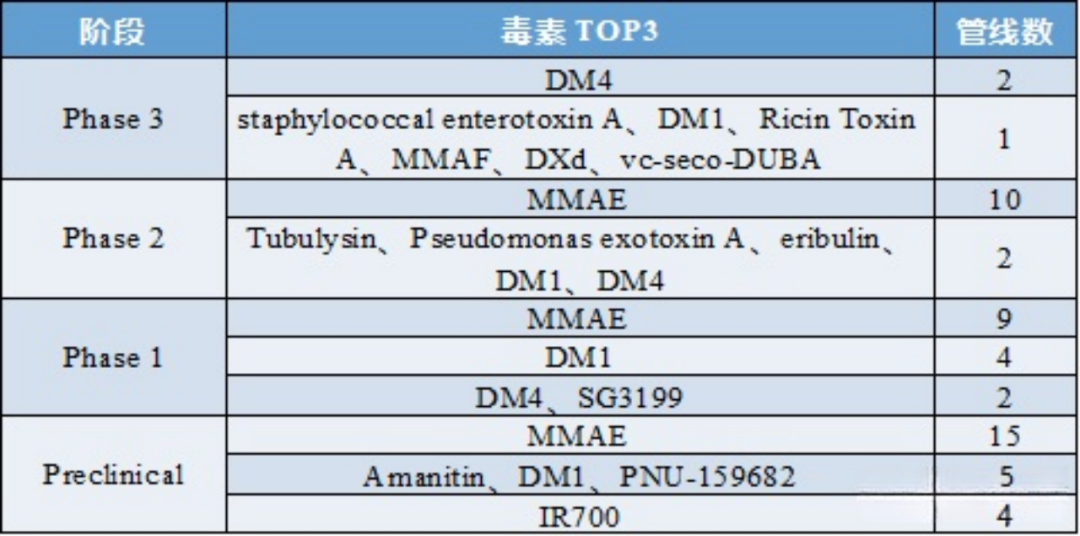
Source: 2022.2 “Overview of ADC Drug Toxins and Linkers”
Table 4: Competition Landscape and Quantity of TOP20 Toxins in ADC Drug Field
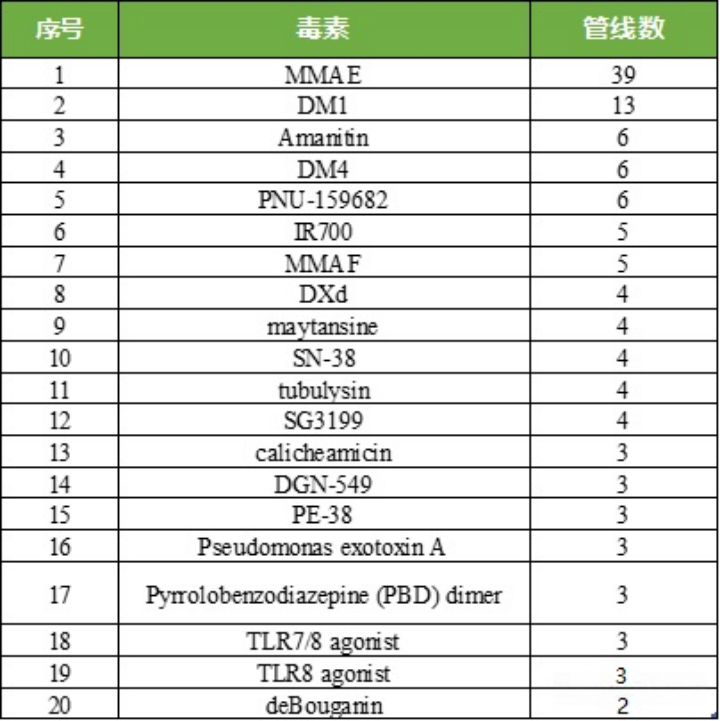
Source: 2022.2 “Overview of ADC Drug Toxins and Linkers”
04 Common Toxin IC50
To address the issue of insufficient ADC efficacy, injecting higher doses of drugs is clearly not a feasible approach; exploring the route of using more potent payloads is still worth trying.
With the emergence of more efficient toxins, ADCs are also beginning to upgrade, relying on more potent toxins to significantly enhance their efficacy. The second-generation payloads are primarily microtubule inhibitors, which can interfere with mitosis, as tumor cells divide faster than most normal cells, making microtubule inhibitors more effective against cancer cells. Microtubule inhibitors can be divided into two types, one that promotes microtubule polymerization and allows uncontrolled microtubule growth, such as the auristatin derivatives MMAE and MMAF; the other effectively inhibits microtubule assembly, inducing mitotic arrest of cells, such as the maytansine derivatives DM1 and DM4. Microtubule inhibitors are currently the most mature payloads, with toxicity significantly stronger than traditional chemotherapy drugs, but there are also issues, such as microtubule inhibitors only being able to kill dividing tumor cells and being ineffective against non-dividing and static cancer cells, leading to resistance.
In contrast, DNA inhibitors, which can act throughout the entire cell cycle, not only kill non-dividing cancer cells but also target cancer cells resistant to classic microtubule inhibitors. As third-generation payloads, DNA inhibitors can destroy DNA structure and function through double-strand breaks, alkylation, embedding, cross-linking, and inhibiting topoisomerase I (TOPO1), promoting cancer cell death.
Compared to the nanomolar IC50 (half-maximal inhibitory concentration) ranges observed in microtubule inhibitors, DNA-damaging agents can achieve IC50 values at the picomolar level, making ADCs combined with DNA-damaging agents sometimes more effective and potentially independent of the cell cycle (compared to microtubule inhibitors that primarily act on dividing cells), and they may even be used on cells with low antigen expression. The common IC50 values of various toxins are shown in the table below.
Table 5: IC50 Range Values of Common Toxin Types
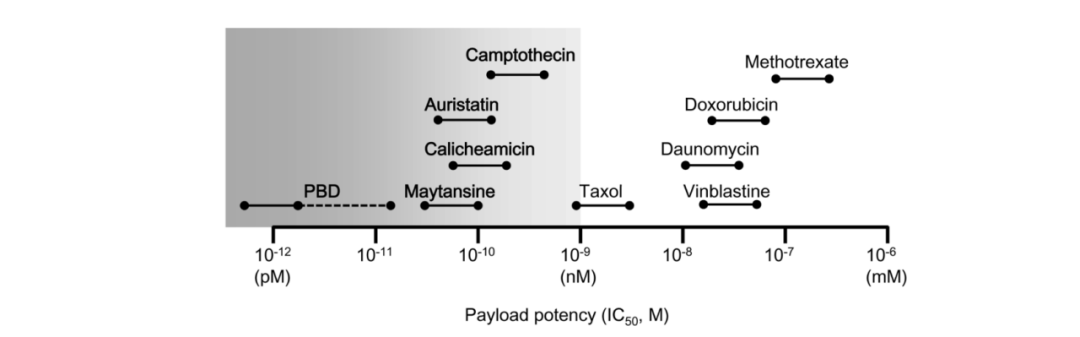
Source: 2018, The Latest Research and Development into the Antibody–Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy
Table 6: Activity of Specific Toxin Molecules (IC50)
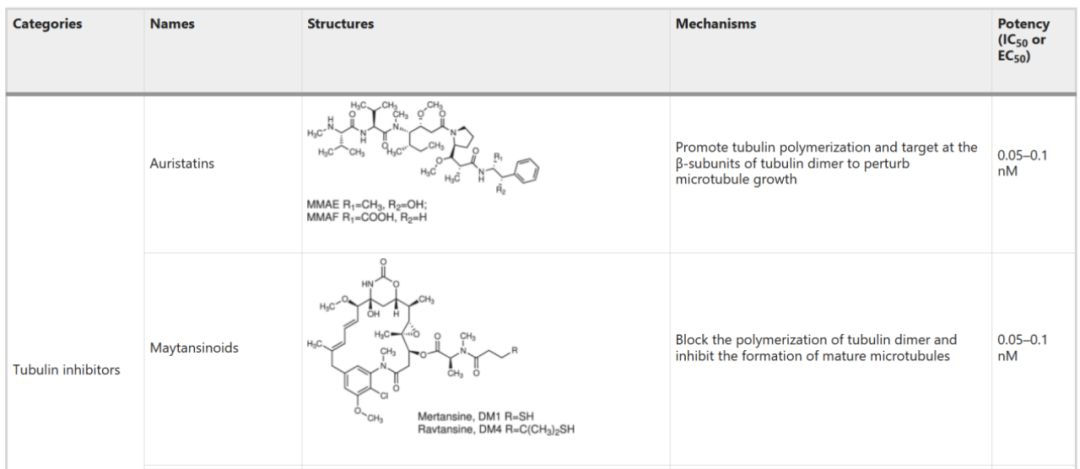
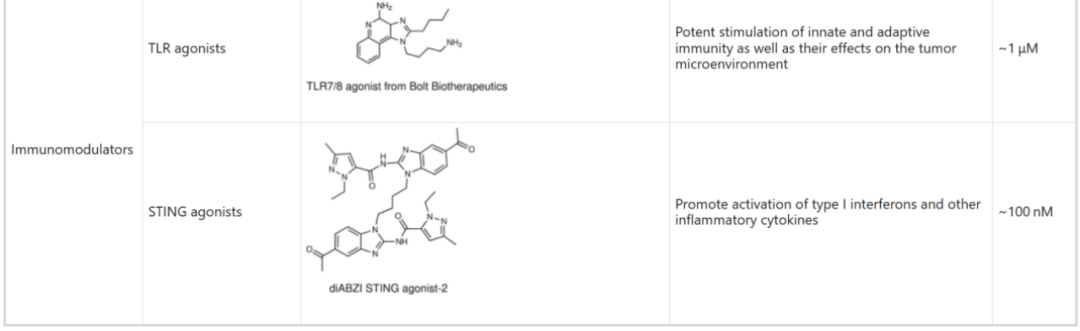
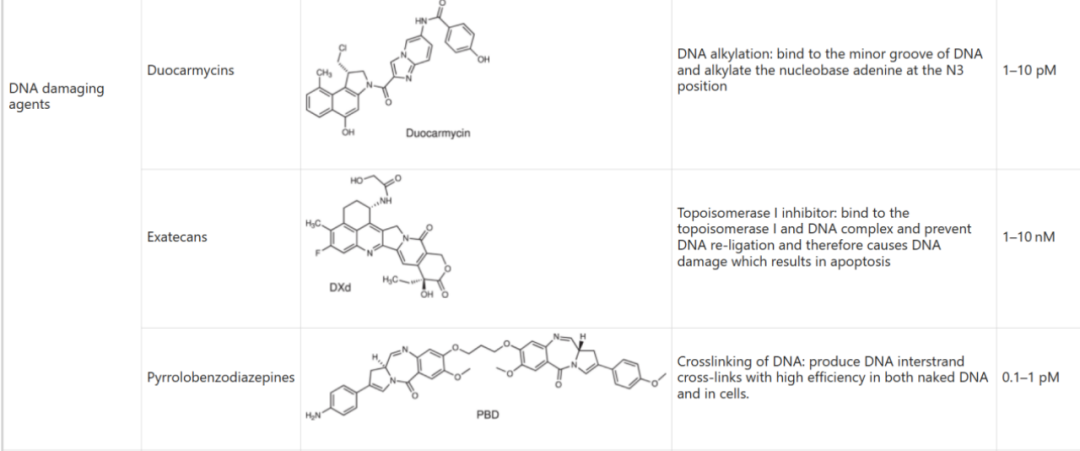
Source: 2022, Antibody drug conjugate: the “biological missile” for targeted cancer therapy
References:[1] 2022, Zhiwen Fu, Antibody drug conjugate: the “biological missile” for targeted cancer therapy[2] 2018, Takashi Nakada, The Latest Research and Development into the Antibody–Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy[3] 2022, Guo Zuhao, Wang Sihan, Overview of Payloads in ADC Drug Development[4] 2023, Liu Wenchao; Li Hongfeng; Hu Zhaohong, Current Status and Prospects of Antibody-Drug Conjugates[5] 2022, Kelaiying News, Overview of ADC Drug Toxins and Linkers
Service:
This public account accepts free submissions of research progress, development stories, etc., from research teams/units for non-commercial/non-profit purposes, and also offers free postings of recruitment ads for research teams. Please contact us through the backend.
Disclaimer: Publishing/reproducing this article is solely for the purpose of disseminating information and does not represent the views of this public account or verify the authenticity of its content. Any judgments made based on this content are at your own risk.If there is any infringement, please inform us for deletion!
Long press to follow this public account
 Fan Group/Submission/Authorization/Advertisement etc.Please contact the public account assistant
Fan Group/Submission/Authorization/Advertisement etc.Please contact the public account assistant  If you find this article interesting, please click here↓
If you find this article interesting, please click here↓