Organic light-emitting diodes (OLEDs) technology has deeply penetrated the fields of display and lighting, with its core relying on the continuous innovation of organic luminescent materials. From the initial use of traditional fluorescent materials that could only utilize 25% of singlet excitons, to phosphorescent materials and thermally activated delayed fluorescence (TADF) materials that can utilize triplet excitons through heavy metal effects or reverse intersystem crossing (RISC), the internal quantum efficiency of devices can theoretically reach 100%. However, facing more cutting-edge applications such as electrically pumped organic lasers, material development still faces severe challenges: the stability of high-performance blue phosphorescent materials is insufficient, the charge transfer state characteristics of TADF materials lead to limited computational accuracy, and the traditional “trial-and-error” experimental development cycle is long and costly.
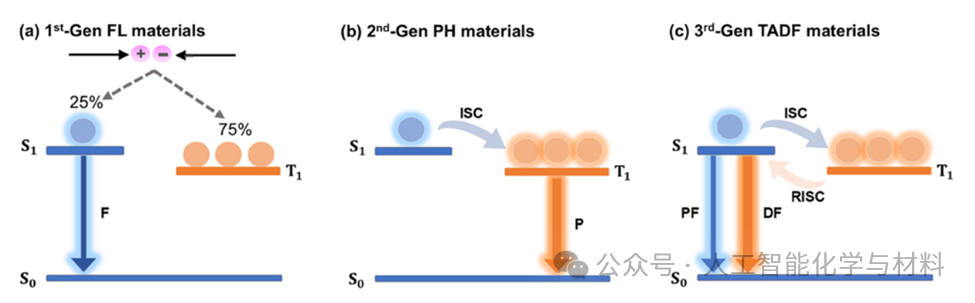
Figure 1. Schematic diagram of the luminescence mechanism of organic luminescent materials.This figure clearly compares the luminescence pathways of three generations of OLED materials. The first generation of fluorescent materials emits light only through the radiative transition of singlet excitons, with an internal quantum efficiency limit of 25%. The second generation of phosphorescent materials utilizes the strong spin-orbit coupling of heavy metal atoms, making the originally forbidden radiative transition from triplet to ground state possible, thus achieving 100% internal quantum efficiency. The third generation of TADF materials converts triplet excitons to singlet excitons through thermally activated reverse intersystem crossing, thereby emitting delayed fluorescence, with its efficiency core being the extremely small singlet-triplet energy gap. This figure is fundamental to understanding the physical basis of the research subject throughout the article.(Image source: Chem Soc Rev)
In this context, theoretical calculations and simulations play an increasingly critical role. The team led by Professor Shuai Zhigang from Tsinghua University / The Chinese University of Hong Kong (Shenzhen) has long been committed to developing computational methods for excited state structures and dynamics. Their research aims to build a “predictable and interpretable” bridge that connects microscopic molecular structures with their macroscopic optoelectronic properties. This article will delve into how the team promotes organic luminescent materials into a new stage of “rational design” through the deep integration of quantum chemistry, machine learning, and rate theory.

This article focuses on the cutting-edge applications of AI in the theoretical design and screening of efficient organic luminescent materials (including fluorescent, thermally activated delayed fluorescence, and phosphorescent materials). The research team has constructed a precise prediction and high-throughput virtual screening system from molecular structure to photophysical properties (such as luminescence quantum yield, laser gain, and emission spectrum) by integrating quantum chemistry calculations, self-developed rate theory software MOMAP, and a three-dimensional molecular representation learning model Uni-Mol. This cross-scale computational strategy has been successfully applied to the screening of candidate molecules for organic lasers, spectral prediction of phosphorescent complexes, and automated design, providing a new paradigm for solving the “accuracy–efficiency” trade-off dilemma in material development. Related methodologies and case studies have been published in Chemical Society Reviews (DOI: 10.1039/d5cs00959f).
Method and Technology Innovations
The core technical route of the research team is established on the close coupling of three major pillars: strictly benchmarked quantum chemistry methods, self-developed photophysical property calculation software MOMAP, and machine learning models for three-dimensional molecular structures Uni-Mol.
In selecting quantum chemistry methods, the team did not seek a “one-size-fits-all” solution, but instead constructed a hierarchical “decision tree” based on the photophysical mechanisms of the material systems. For conventional fluorescent small molecules, methods such as B3LYP, PBE0, and other functionals of TD-DFT can achieve a good balance between accuracy and efficiency. When facing TADF molecules with significant charge transfer characteristics, traditional functionals fail due to poor description of CT states, necessitating a shift to long-range corrected functionals such as ωB97XD, CAM-B3LYP, or tuning ω to accurately capture the key parameters determining RISC efficiency—namely, the singlet-triplet energy gap. For cutting-edge multi-resonance TADF molecules, their excited states contain significant double excitation features, rendering most TD-DFT methods inapplicable, necessitating the use of wave function methods that include higher-order excitations, such as SCS-CC2, ADC(2), or double hybrid functionals, despite significantly increased computational costs. This physics-based methodological choice is a prerequisite for achieving accurate predictions.
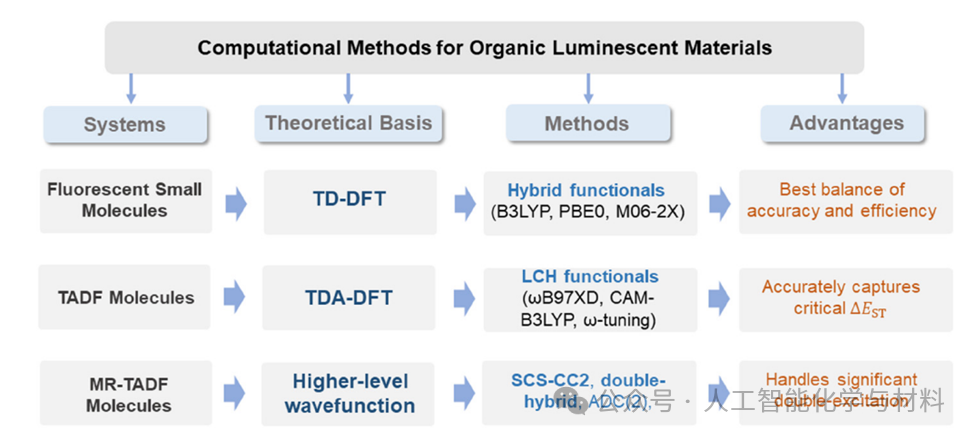
Figure 2. Decision tree for selecting computational methods for organic luminescent materials.This figure vividly illustrates the computational method selection strategy for different luminescent systems. It emphasizes the core idea that “there is no universally applicable computational method.”The decision process begins with understanding the luminescence mechanism of the molecular system: is it localized excitation, charge transfer, or multi-resonance effects? Based on this, researchers are guided to the most suitable quantum chemistry methods, from computationally inexpensive TD-DFT to more accurate but time-consuming wave function methods. This decision tree is an important tool for guiding researchers in reliable calculations. (Image source: Chem Soc Rev)
After obtaining accurate electronic structures, the team uses the self-developed MOMAP software package to calculate photophysical properties. MOMAP is based on the theory of thermal vibration correlation functions, comprehensively considering mode displacements and Duschinsky rotation effects between different potential energy surfaces under harmonic approximation, enabling the calculation of radiative rates, non-radiative rates (including internal conversion, intersystem crossing, and their reverse processes), as well as absorption and emission spectra. This method avoids the shortcomings of traditional displacement harmonic oscillator models that neglect mode rotation, and all input parameters can be obtained from mainstream quantum chemistry software without any empirical parameters, thus achieving parameter-free, quantitative predictions of key performance indicators such as photoluminescence quantum yield.
To achieve high-throughput exploration of vast chemical space, the team introduced the Uni-Mol three-dimensional molecular representation learning model. Unlike traditional models based on one-dimensional sequences or two-dimensional graph structures, Uni-Mol directly takes the three-dimensional coordinates of molecules as input, efficiently learning the three-dimensional geometry and electronic structure information of molecules by introducing relative position encoding and bidirectional information exchange between atom-atom pairs. After pre-training on a large amount of unlabelled data and fine-tuning on specific properties, Uni-Mol can quickly and accurately predict HOMO/LUMO energy levels, excitation energies, and even complex PLQY, significantly improving screening efficiency by several orders of magnitude.
Main Findings and Result Analysis
In the screening of organic laser materials, the team established a rigorous computational screening protocol. They first verified the good linear relationship between theoretical predictions and experimental laser thresholds through systematic calculations of 12 known laser molecules and 23 non-laser molecules. The core of this protocol is to calculate the net stimulated emission cross-section under optical pumping and electrical pumping, the latter of which requires additional deduction of absorption losses caused by triplet and polaron states. Using this standard, they successfully screened potential electrically pumped laser candidate molecules from known fluorescent molecules, including BP3T, CzPVSBF, and BSBCz, among which BSBCz’s electrically narrow spectrum emission has been experimentally confirmed, validating the effectiveness of the protocol.
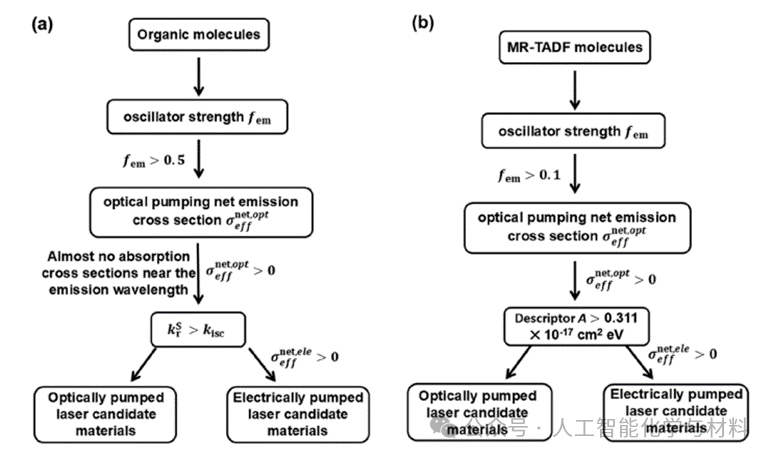
Figure 3. Diagram of theoretical prediction and screening scheme for organic laser molecules.Figure 3a summarizes the general computational screening process for organic small molecule laser materials, emphasizing the derivation of key photophysical parameters (such as oscillation strength, radiative/non-radiative rates, various absorption/emission cross-sections) from quantum chemistry calculations, ultimately screening candidate molecules through the relationship between net gain cross-section and laser threshold. Figure 3b specifically targets MR-TADF molecules, demonstrating how to achieve rapid initial screening using theoretical descriptors A, greatly enhancing screening efficiency. These two flowcharts clearly outline the complete path from theoretical calculations to material design.(Image source: Chem Soc Rev)
Expanding this protocol to TADF and MR-TADF systems, the team evaluated 21 molecules. Through stepwise screening (eliminating molecules with excessively low oscillation strength, rapid triplet non-radiative rates, severe self-absorption, or negative net electrical pumping gain), they ultimately identified DABNA-2, m-Cz-BNCz, and four other MR-TADF molecules as highly promising electrically pumped laser candidate materials. Notably, the laser behavior of DABNA-2 and another candidate molecule 4CzTPN has been validated by subsequent experiments, further proving the predictive nature of this computational screening strategy.
To further accelerate the discovery of MR-TADF laser materials, the team proposed a concise theoretical descriptor A, defined as the product of the singlet-triplet energy gap and the net stimulated emission cross-section under optical pumping. By calculating 119 virtual design MR-TADF molecules, they found that descriptor A effectively narrowed the candidate range, quickly locking in 8 potential molecules, among which ADBNA-Me-BPy is considered a star molecule for achieving low-threshold electrically pumped lasers due to its extremely high radiative rate and RISC rate.
In the field of phosphorescent materials, the team conducted comprehensive computational benchmarking on 50 types of four-coordinate platinum (II) complexes. They compared the performance of 14 types of density functionals under four different definitions of emission energy, finding that the B3LYP*/E_ad,u combination predicted emission energy most accurately. In terms of spectral shape prediction, functionals such as TPSSh, TPSS, BP86, PBE, and B3LYP* exhibited the best robustness. By analyzing charge density differences and reorganization energy distributions, they revealed the intrinsic correlation between molecular structure and color purity: for molecules achieving narrow spectral emission, the regions of electronic density change highly coincide with the dominant vibrational modes; while the extension of conjugated structures activates more low-frequency vibrational modes, leading to spectral broadening.
The most remarkable progress was made by the team utilizing the Uni-Mol model to achieve automated high-throughput screening of iridium (III) complexes. Starting from about 278 bidentate ligands, they constructed a virtual library containing 1.6 million candidate molecules. After pre-optimization based on GFN2-xTB and pre-training and fine-tuning of Uni-Mol, the model could quickly predict the molecules’ HOMO/LUMO energy levels, T1 state adiabatic excitation energies, and PLQY. The final screening process combined emission color and PLQY thresholds, successfully “retrieving” multiple known high-performance phosphorescent materials and newly discovering several candidates with narrow full width at half maximum, covering from blue light to red light and even achieving pure white emission, fully demonstrating the powerful capability of AI-driven material design.
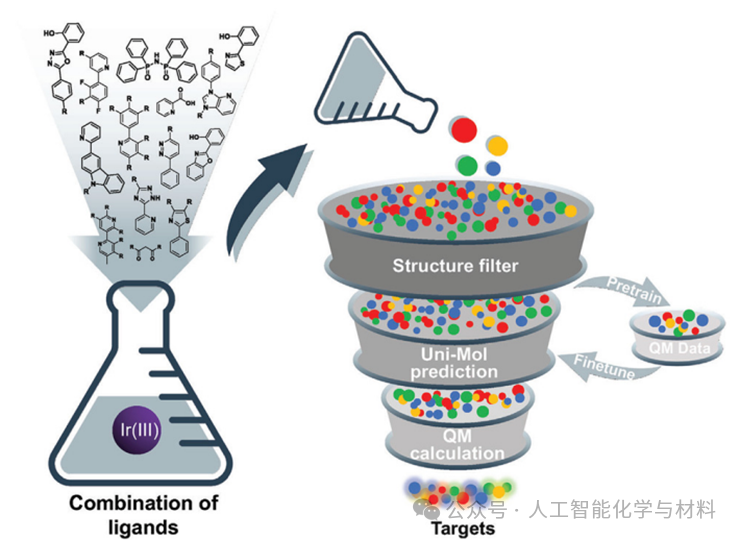
Figure 4. Workflow diagram for high-throughput virtual screening of Ir(III) complexes.This figure details the automated screening pipeline based on Uni-Mol. From ligand library construction, virtual molecule generation, to pre-screening based on GFN2-xTB, pre-training and property prediction fine-tuning of Uni-Mol, and finally multi-level screening based on color and PLQY and other performance indicators, the entire process is fully automated. This figure is a model of the AI-driven new material discovery process, demonstrating how to seamlessly integrate computational chemistry, big data, and machine learning to explore vast chemical space.(Image source: Chem Soc Rev)
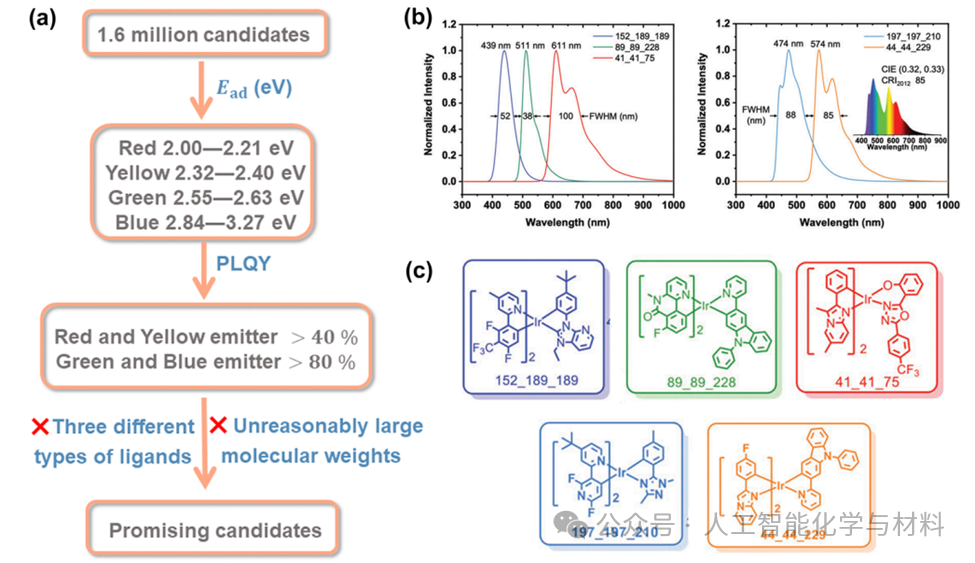
Figure 5. Selected Ir(III) candidate molecules and their emission spectra.Figures 5b and 5c show the chemical structures and predicted emission spectra of five representative iridium (III) complexes screened through the aforementioned HTVS process. The results indicate that two candidate molecules (green emitter 89_89_228 and blue emitter 152_189_189) exhibit extremely narrow full width at half maximum, indicating high color purity. Interestingly, by adjusting the ratio of cyan emitter 197_197_210 and yellow-orange emitter 44_44_229, pure white light with ideal coordinates can theoretically be obtained. These results not only validate the effectiveness of the screening process but also provide a direct design blueprint for its application in next-generation display technologies.(Image source: Chem Soc Rev)
References and Index
Original paper: Combining quantum chemistry, machine learning and rate theory for organic luminescent materials. Chem. Soc. Rev., 2025, Advance Article
https://doi.org/10.1039/D5CS00959F
 Disclaimer:This article is for academic exchange among researchers only. If there are any copyright issues or questions, please contact us for timely resolution. Contact email:[email protected]
Disclaimer:This article is for academic exchange among researchers only. If there are any copyright issues or questions, please contact us for timely resolution. Contact email:[email protected]