High-throughput sequencing technology is driving the life sciences towards a quantitative paradigm, but the sequencing technologies for antibodies, glycans, and other non-directly measurable molecules face challenges such as complex coupling steps, steric hindrance, and the labor-intensive nature of enzymatic chemical labeling. Aptamers, as nucleic acid molecules that can be sequenced directly, possess high specificity, broad targeting capabilities, and small size advantages, making them ideal tools for multi-modal molecular analysis. This study proposes the Apt-seq platform, a high-throughput single-cell multi-omics quantitative technology based on aptamers. This integrated strategy, termed “aptameromics,” allows for the parallel analysis of cell surface proteins, glycans, and mRNA. Validation in commercial cell lines shows that sequencing data is highly consistent with flow cytometry results at both the population and single-cell levels. When applied to clinical samples, Apt-seq can accurately resolve tumor heterogeneity, identifying a subpopulation of tumor stem cells with high PTK7 expression, confirming its potential as a stem cell marker. Additionally, by targeting sialic acid with aptamers, dynamic changes in sialylation during T cell differentiation were tracked. Apt-seq achieves simultaneous quantification of directly and indirectly measurable molecules, providing a universal scalable platform for comprehensive quantitative analysis of biomolecules in complex biological systems, thus advancing the development of quantitative science.


Results Analysis

1.Apt-seq Workflow.
This study developed the Apt-seq technology, a method for high-throughput quantitative analysis of biomolecules such as mRNA, proteins, and glycans at the single-cell level (Figure 1). The research utilized the 10× Genomics single-cell RNA-seq platform, which employs a droplet-based system to digitally count 3′ end mRNA from thousands of single cells. The authors extended the aptamers with universal PCR handles and sequences complementary to the capture sequences on single-cell microbeads, allowing seamless integration with this platform. This design ensures independent capture of aptamers and mRNA, avoiding mutual interference and maintaining the accuracy of sequencing data. Experimental validation showed that the extended sequences do not affect the specificity of aptamer binding, allowing for independent capture and amplification of biomolecules during library construction, thus avoiding cross-contamination. After sequencing, bioinformatics analysis clusters cells and molecular features, providing multidimensional data for resolving cellular heterogeneity.
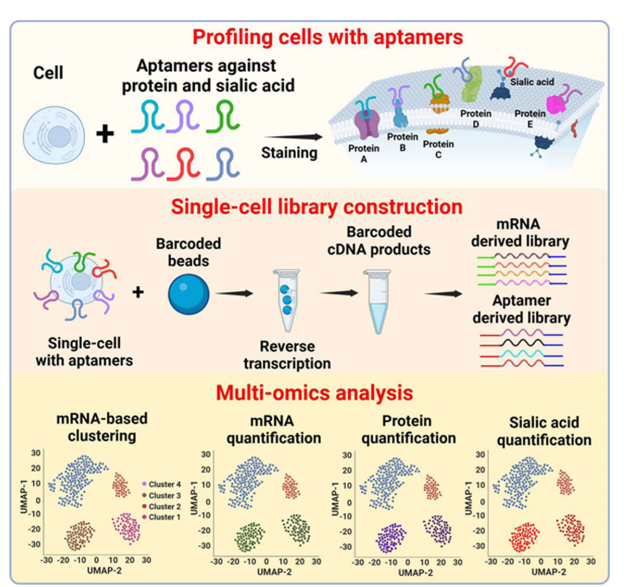
2.Apt-seq Technology for Analysis of Cell Surface Proteins at Bulk Level
The study first compared the binding trends of aptamers targeting CD71 with antibodies across six cell types, finding a strong positive correlation (r=0.8946, p<0.05), confirming that aptamers can replace antibodies for cell surface protein analysis. Subsequently, 15 distinct aptamers were used to analyze 20 cell lines, achieving precise quantitative sequencing through the introduction of unique molecular identifiers (UMIs), and compared with flow cytometry. The results showed strong positive correlations between sequencing and flow data for the suspension cell line WSU DLCL-2 (r=0.8254, p<0.0005) and the adherent cell line MGC-803 (r=0.9485, p<0.0001), indicating that Apt-seq is a reliable high-throughput detection method. Additionally, a highly enriched aptamer library (R16) targeting the ovarian cancer cell line OVCAR-4 was obtained through 16 rounds of cell-SELEX, and flow analysis showed its ability to distinguish 13 cell lines. NGS analysis indicated that the coefficient of variation (CV) for four independent sequencing runs was <15%, with a Spearman correlation coefficient >0.97, demonstrating high stability and reliability. Clustering analysis revealed that 500 aptamers in the library could distinguish different cell lines based on unique binding patterns, and the binding differences with OVCAR-3 and OVCAR-4 were consistent with the known molecular features of the two cell lines, validating the effectiveness of aptamers in recognizing cellular surface molecular heterogeneity and providing a new method for multi-parameter cell characterization.
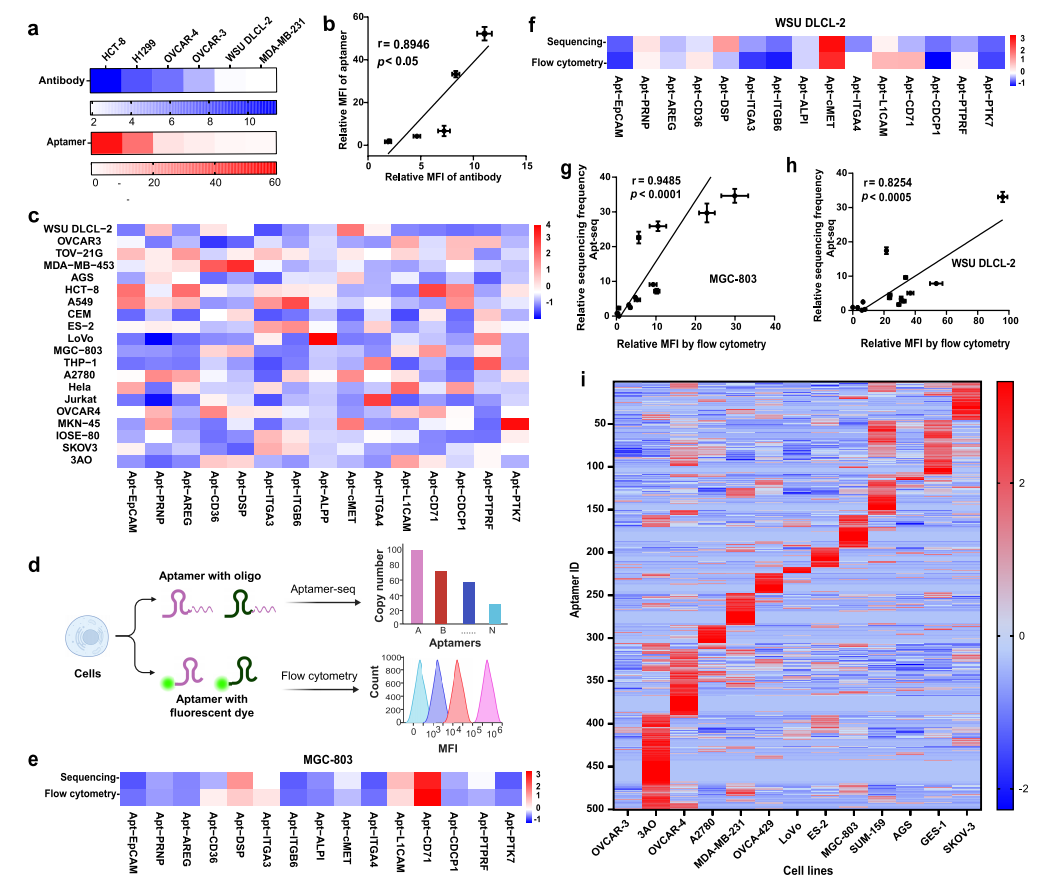
3.Apt-seq Technology for Analysis of Cell Surface Proteins at Single-Cell Level
To validate the applicability of Apt-seq at the single-cell level, the study first compared single-cell sequencing data from cells with and without aptamer incubation, finding a high correlation in gene expression (r=0.98, p<2.2×10⁻¹⁶), confirming that aptamer treatment does not affect the cellular transcriptome. Subsequently, titration experiments were conducted using 5-500 nM of apt-PTK7 on CCRF-CEM cells, with single-cell sequencing showing a dose-dependent binding, reaching a plateau at 200 nM, with an apparent dissociation constant (25.4 nM) consistent with flow cytometry (26.7 nM). Comparison with CITE-seq indicated that the sequencing reads of the aptamer targeting CD49 were comparable to those of the antibody-oligonucleotide complex, validating the accuracy of Apt-seq. This technology was applied to analyze clinical samples from ovarian cancer, with transcriptome UMAP analysis identifying epithelial tumor cells, mesenchymal stem cells, and fibroblasts. Among them, the abundance of aptamers targeting EpCAM and PTK7 was significantly correlated with their corresponding mRNA, and their distribution was consistent with the expression characteristics of cell types. Of the 13 aptamers, 10 showed positive correlation with their target proteins, while 3 showed no correlation due to expression differences. This study confirms the reliability of Apt-seq for quantitative analysis at the single-cell level, as well as its advantages in resolving cellular heterogeneity and directly quantifying proteins in complex solid tumor samples.
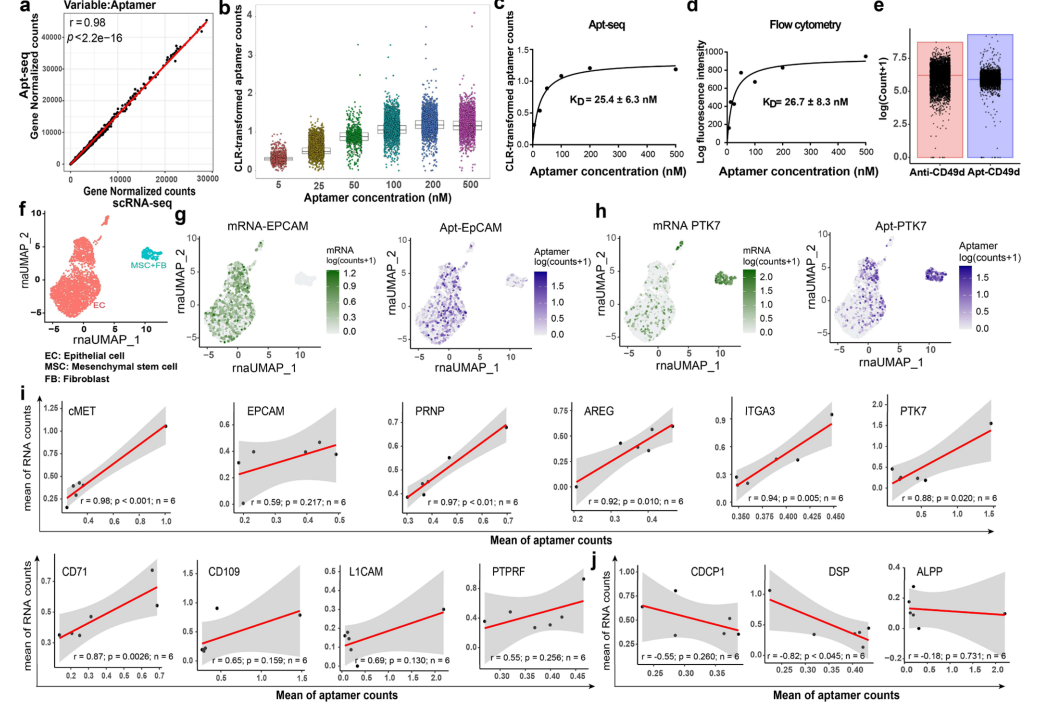
4.Apt-seq Enables Sensitive Analysis of Cellular Heterogeneity in Clinical Hematological Samples
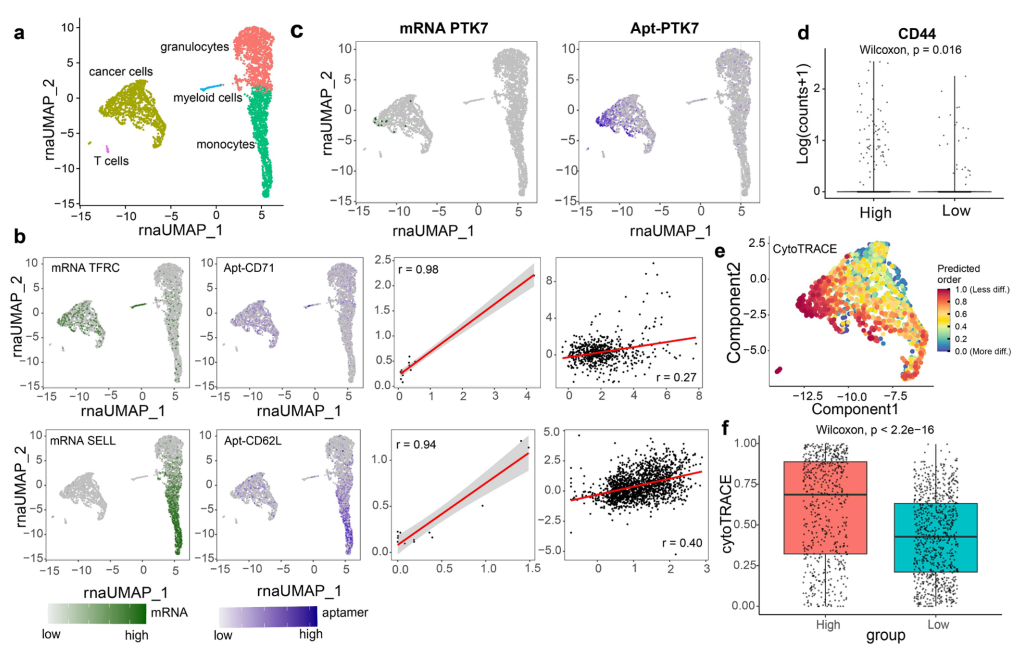
After validating the applicability of Apt-seq in clinical solid tumor samples, the study further assessed its feasibility in clinical blood samples using bone marrow samples from newly diagnosed multiple myeloma (MM) patients. UMAP analysis based on mRNA expression showed the presence of granulocytes, myeloid cells, monocytes, T cells, and a large number of tumor cells in the samples. Aptamers targeting CD71 and SELL primarily localized to tumor/myeloid cells and monocytes, respectively, and were significantly correlated with their corresponding mRNA expression, confirming the reliability of Apt-seq. Aptamers were found to be more sensitive than mRNA sequencing in detecting cell markers, especially for low-expressing mRNAs. For instance, the distribution of the PRNP aptamer in cancer cells far exceeded that of PRNP mRNA expression. Furthermore, the study identified a subpopulation of tumor cells in MM samples with strong binding of PTK7 aptamers, which exhibited higher expression of the stem cell marker CD44, and CytoTRACE analysis indicated stronger stemness, while distinguishing this subpopulation based on PTK7 mRNA was challenging. These results suggest that Apt-seq combined with multi-omics analysis can effectively reveal tumor cell heterogeneity, providing a more sensitive tool for cell phenotype identification.
5.Multi-Modal Analysis of Transcriptome, Cell Surface Proteins, and Cell Surface Metabolites in Clinical Blood Samples
Aptamers have a broader target recognition range than antibodies, and Apt-seq enables multi-modal analysis of transcriptomes, cell surface proteins, and metabolites. In the experiment, aptamers targeting sialic acid were added, and their binding to cells could be weakened by neuraminidase treatment, confirming specificity (Figure S10). Analysis of bone marrow samples from myeloma treated with IFN-γ showed that Apt-seq could identify various cell populations, including B cells and T cells, and the number of aptamer detections per single cell was higher than that of mRNA, which is related to the low abundance detection limitations caused by mRNA diversity. After IFN-γ treatment, the mRNA levels of molecules such as PD-L1 changed in accordance with aptamer signals, while others like ITGA4 exhibited expression differences, reflecting the complexity of spatiotemporal regulation between mRNA and proteins. Additionally, IFN-γ enhanced the binding ability of sialic acid aptamers, consistent with lectin results in Ramos cells, with B cells showing the highest levels of sialic acid (Figure 5f-h). This study confirms that Apt-seq, with its broad targeting capabilities, provides an effective tool for precise quantification of non-measurable biomolecules, aiding in the in-depth analysis of cellular states.
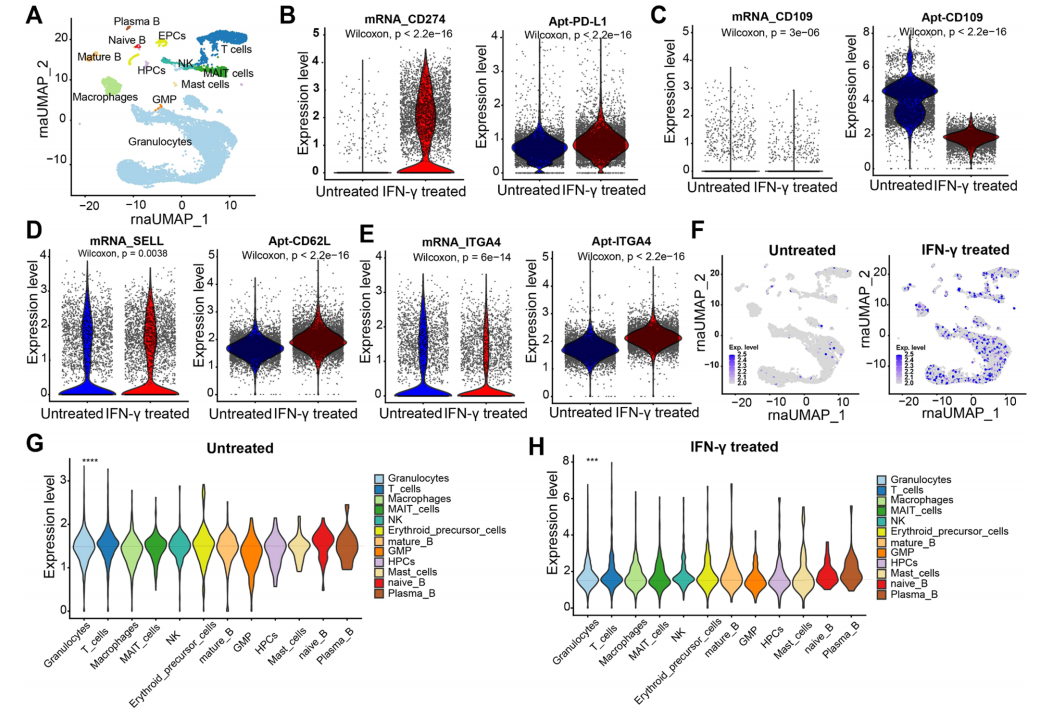
6.Using Apt-seq to Track Changes in Cell Surface Metabolite Levels During T Cell Differentiation
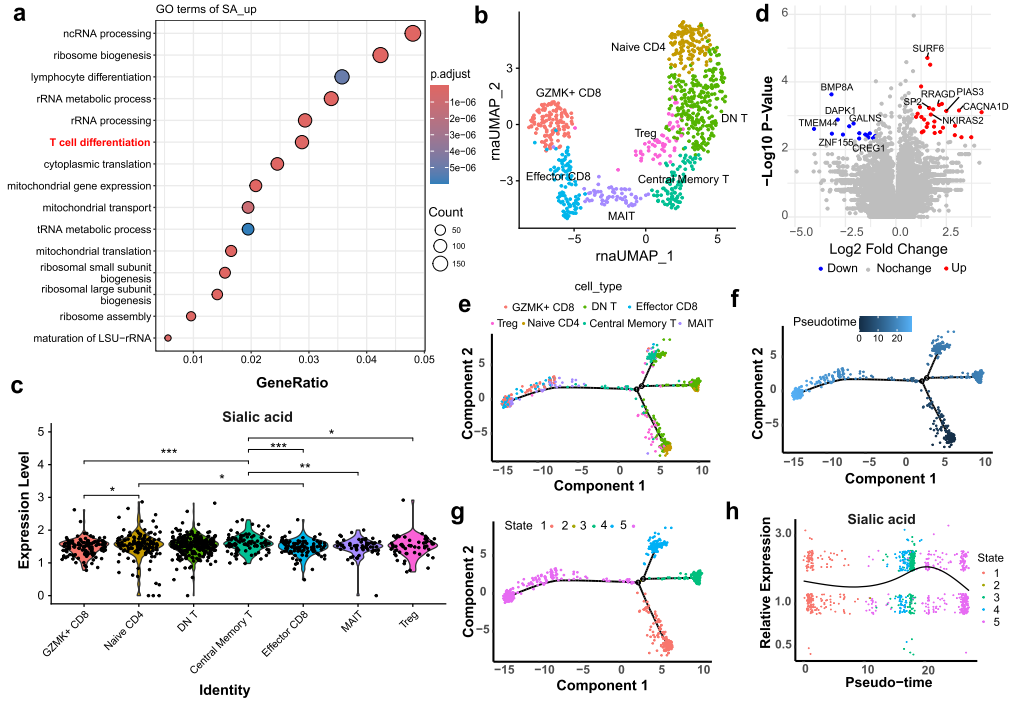
Based on sialic acid aptamer binding counts, untreated multiple myeloma bone marrow cells were divided into “SA_high” and “SA_low” groups, revealing that sialic acid levels were associated with T cell differentiation. After isolating T cells, they were divided into seven subpopulations based on activation stages, showing significant differences in sialic acid levels during differentiation. In the “SA_high” T cell group, genes related to T cell activity, such as PIAS3 and NKIRAS2, were significantly upregulated. Analysis of single-cell pseudotime using the Monocle algorithm showed that sialic acid levels increased during the transition from resting T cells to functional T cells and decreased after maturation. This study confirms that Apt-seq can effectively track multi-modal molecules in complex samples, achieving synchronous quantification and dynamic monitoring of metabolite changes during T cell differentiation, providing important tools for revealing the molecular mechanisms of immune cell activation and function, and demonstrating the advantages of this technology in elucidating the dynamic processes of cell differentiation.
Article link: https://pubs.acs.org/doi/10.1021/jacs.5c01296