Since the first ADC drug Mylotarg® (gemtuzumab ozogamicin) was approved by the FDA in 2000, as of December 2021, a total of 14 ADC drugs have been approved globally for hematological malignancies and solid tumors, and currently, there are over 100 ADC candidates at various stages of clinical trials.
Recently, the journal Signal Transduction and Targeted Therapy published an in-depth review by Huazhong University of Science and Technology Tongji Medical College, which reviewed the history and mechanisms of action of ADCs, and discussed the key components of ADCs and their impact on ADC activity. The review also detailed the approved ADC drugs and other promising candidates (in Phase III clinical trials), discussing current challenges and future prospects for the next generation of ADCs. Today, the WeChat public account drug-conjugates team will share the highlights of this review with readers.

1. Timeline of Important Events in ADC Drug Development and Approval
As early as the early 20th century, Paul Ehrlich first proposed the concept of “magic bullets”, hypothesizing that certain compounds could directly enter specific targets in cells to cure diseases. Theoretically, these compounds should effectively kill cancer cells while being harmless to normal cells.
In 2000, the U.S. Food and Drug Administration (FDA) first approved the ADC drug Mylotarg® (gemtuzumab ozogamicin) for adult acute myeloid leukemia (AML), marking the beginning of the era of ADC targeted cancer therapy. Figure 1 depicts the landmark events in the development of ADC drugs from infancy to maturity over the past century. With the continuous expansion of targets and indications, ADCs are leading a new era of cancer targeted therapy and are expected to replace traditional chemotherapy drugs in the future.
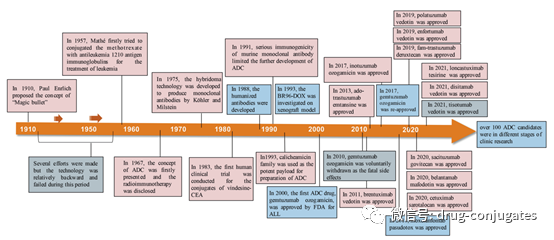
Figure 1 Timeline of Important Events in ADC Drug Development and Approval (Image Source: Reference [1])
1. Composition of ADC Drugs
ADC consists of an antibody, a payload, and a chemical linker. Ideally, ADC drugs remain stable in circulation, accurately reach therapeutic targets, and ultimately release cytotoxic payloads near the target (e.g., cancer cells). Each component affects the final efficacy and safety of the ADC, and overall, ADC development needs to consider all these key components, including targets, antibodies, cytotoxic payloads, linkers, and the choice of conjugation methods.
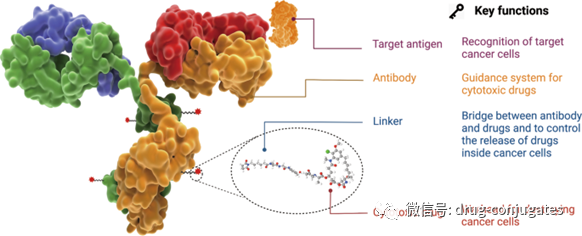
Figure 2 Structure and Characteristics of ADC Drugs (Image Source: Reference [1])
Target Antigens
The target antigens expressed on tumor cells guide ADC drugs in recognizing tumor cells, and they also determine the mechanism by which cytotoxic payloads are delivered to cancer cells (e.g., endocytosis). Therefore, selecting the appropriate target antigen is a primary consideration for ADCs.
Firstly, to reduce off-target toxicity, the targeted antigen should be expressed only or predominantly in tumor cells and not expressed or expressed minimally in normal tissues. For example, HER2 expression in certain types of tumors is approximately 100 times higher than in normal cells, which laid a solid foundation for the development of ado-trastuzumab emtansine, fam-trastuzumab deruxtecan, and disitamab vedotin.
Secondly, the target antigen should be non-secretory, as secreted antigens in circulation can lead to ADC binding outside the tumor site, resulting in reduced tumor targeting and safety issues.
Thirdly, the ideal target antigen should be internalized after binding with the corresponding antibody, so that the ADC-antigen complex can enter the cancer cell and then release the cytotoxic payload through appropriate intracellular transport pathways.
Currently approved ADC drugs typically target specific proteins overexpressed on cancer cells, including HER2, Trop2, Nectin4, and EGFR in solid tumors, as well as CD19, CD22, CD33, CD30, BCMA, and CD79b in hematological malignancies. With the advancement of oncology and immunology research, the selection of ADC target antigens has gradually expanded from traditional tumor cell antigens to targets in the tumor microenvironment (e.g., in the stroma and vascular system). New evidence from preclinical and clinical studies suggests that components of the neovascular system, subendothelial extracellular matrix, and tumor stroma may be valuable target antigens for ADC drug development.
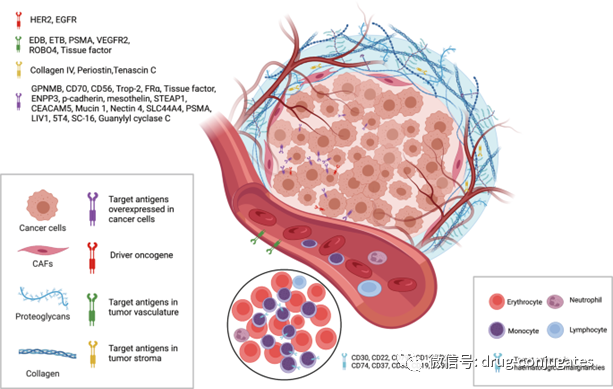
Figure 3 Available Tumor Cell and Tumor Microenvironment (Vascular System and Stroma) Targets for ADCs (Image Source: Reference [1])
Antibodies
Tumor-targeting antibodies are crucial for the specific binding between the target antigen and the ADC. In addition to having high binding affinity to the target antigen, the ideal antibody portion should also promote effective internalization, exhibit low immunogenicity, and maintain a longer plasma half-life.
In the early stages of ADC drug development, mouse-derived antibodies were primarily used, but due to severe immunogenicity-related side effects, the failure rate was high. With the advent of recombinant technology, mouse antibodies have mostly been replaced by chimeric antibodies and humanized antibodies. Currently, ADCs increasingly adopt fully humanized antibodies with significantly reduced immunogenicity. Among the 14 approved ADC drugs, only Brentuximab vedotin used a chimeric antibody.
As the main component of immunoglobulin in serum, the antibodies used in ADC drugs are mostly immunoglobulin G (IgG) antibodies, including four subclasses: IgG1, IgG2, IgG3, and IgG4. IgG1 is the most commonly used subclass in ADCs because it is the most abundant in serum.
The efficiency of internalization of the antibody-antigen complex primarily depends on the binding affinity between the antibody and the tumor cell surface antigen, higher affinity usually leads to faster internalization. However, highly affine antibodies may reduce the penetration of antibodies into solid tumors. Treating solid tumors is more complicated than hematological tumors because there are binding site barriers (BSB) in solid tumors, and extremely high affinity between antibodies and antigens can cause ADCs to become trapped near blood vessels after binding, but penetrate less into tumor cells far from the vessels. Therefore, the reasonable affinity between the antigen and antibody should be optimized to balance the rapid uptake by target cells and anticancer efficacy.
In addition to binding affinity, another factor affecting tumor penetration is the size of the antibody. The large molecular weight (approximately 150 kDa) of IgG antibodies usually presents a significant barrier to penetrating capillaries and the stroma in tumor tissues. Therefore, early ADC drugs primarily targeted hematological malignancies. To enable ADCs to better treat solid tumors, researchers have attempted to miniaturize antibodies by removing the Fc fragment. Miniaturized antibodies not only retain high affinity and specificity but also penetrate blood vessels into solid tumors more easily, greatly enhancing their efficacy against solid tumors. However, it has also been found that this change can lead to a reduced half-life. Therefore, various factors should be considered when designing ADCs with miniaturized antibodies.
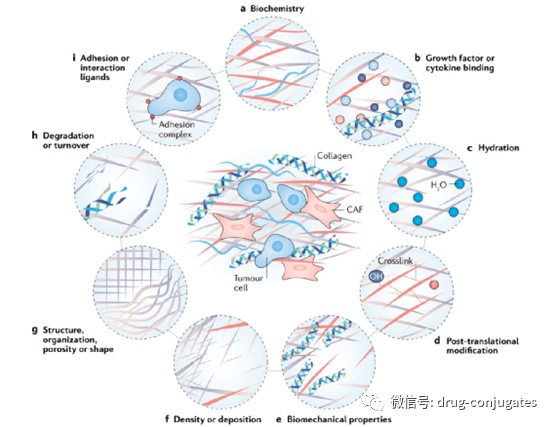
Figure 4 Changes in Stroma in Cancer (Image Source: Reference [2])
Linkers
The linker in ADC connects the antibody to the cytotoxic drug. It has a significant impact on the stability of the ADC and the release profile of the payload, which is crucial for the final efficacy of the ADC drug. The ideal linker should not induce ADC aggregation, prevent premature release of the payload in plasma, and effectively release it inside cancer cells. Depending on the metabolic fate in the cell, most ADC drugs use two types of linkers, including cleavable and non-cleavable linkers.
Cleavable linkers utilize the environmental differences between systemic circulation and tumor cells to accurately release free cytotoxic drugs, which can be further divided into chemical cleavable linkers (hydrazone bonds and disulfide bonds) and enzyme-cleavable linkers (glucosidic bonds and peptide bonds). Another type of cleavable linker is chemically sensitive to reducing glutathione (GSH). GSH plays a crucial role in maintaining intracellular redox balance during cell survival, proliferation, and differentiation. The concentration of GSH in the blood is far lower than its intracellular concentration in cancer cells. Therefore, this type of linker can remain stable in the bloodstream while specifically releasing active payloads inside cancer cells.
Peptide-based linkers are sensitive to lysosomal proteases and have been used in many ADCs. Lysosomal proteases, such as cathepsin B, are often overexpressed in cancer cells, allowing for accurate drug release near the tumor. Additionally, due to the presence of protease inhibitors in the blood, enzyme-cleavable linkers are usually stable in systemic circulation, thus reducing various risks associated with premature cleavage. Among the approved ADC drugs, 9 out of 14 drugs use peptide-based linkers.
Non-cleavable linkers (e.g., thioether or maleimide-based hexanoic acid) are inert to common chemical and enzymatic environments in vivo. The main advantage of non-cleavable linkers is their increased plasma stability, leading to lower off-target toxicity, but their bystander effect on the payload is affected.
Payloads
The payload is the cytotoxic warhead that exerts cytotoxicity after ADC is internalized into cancer cells. Since only about 2% of ADC can reach the target tumor site after intravenous administration, the compounds used as payloads in ADCs need to be highly toxic (IC50 in the nM and pM range). Furthermore, these compounds should remain stable under physiological conditions and have functional groups that can covalently bind with antibodies. Currently, the cytotoxic payloads used in ADCs mainly include potent microtubule inhibitors, DNA damaging agents, and immune modulators.
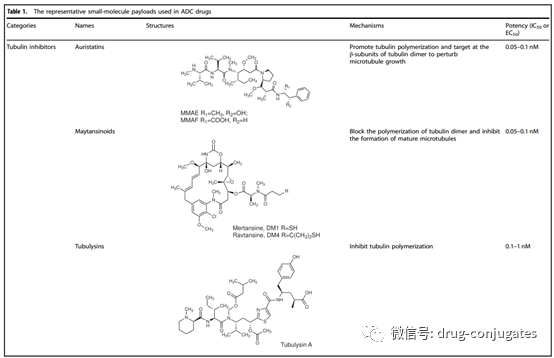
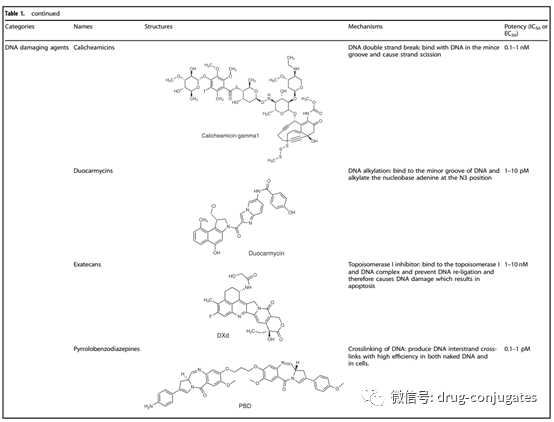
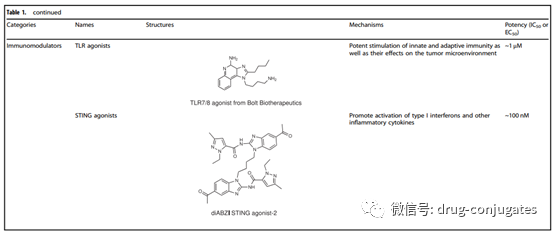
Figure 6 Representative Small Molecule Payloads Used in ADC Drugs (Image Source: Reference [1])
Conjugation Methods
In addition to selecting antibodies, linkers, and payloads, the method of connecting the small molecule part (i.e., linker plus payload) to the antibody is also crucial for successfully constructing ADCs. Typically, the presence of lysine and cysteine residues on the antibody provides reactive sites for conjugation, and early ADC drugs were usually conjugated randomly through lysine or cysteine residues. However, antibodies typically contain about 80-90 lysine residues, of which 40 lysine residues are usually reactive. Random conjugation with lysine residues can lead to a wide distribution of drug-antibody ratios (DAR) with different numbers (0-8) of small molecule toxins attached to the antibody, resulting in a heterogeneous mixture. Additionally, since lysine residues are distributed throughout the light and heavy chains of the antibody, conjugation reactions near the antibody-antigen recognition sites may reduce the binding of the ADC to the target.
Cysteine-based reactions provide another conjugation method. Typically, IgG1 antibodies have both interchain disulfide bonds and intrachain disulfide bonds. Interchain disulfide bonds are exposed externally on the antibody and can be easily reduced to expose free cysteine residues, providing available sites for the conjugation of the linker payload with the antibody. Due to the limited number of binding sites and the unique reactivity of thiol groups, using cysteine as a conjugation site helps reduce the heterogeneity of ADCs. Depending on the reduction rate, products with DAR of 2, 4, 6, and 8 may produce better homogeneity compared to products conjugated with lysine residues. This has become the most commonly used conjugation method in commercial products to date. However, it is worth noting that opening interchain disulfide bonds may compromise the integrity of the antibody.
Random conjugation of lysine and cysteine residues may lead to several issues. The stability of this conjugation is sometimes insufficient, which can lead to premature release of the payload, resulting in off-target toxicity. Additionally, it is challenging to ensure that the payload is connected to consistent sites on the antibody, increasing the difficulty of controlling quality and uniform DAR values. To reduce the heterogeneity of ADCs, several site-specific conjugation strategies have been developed in new ADCs.
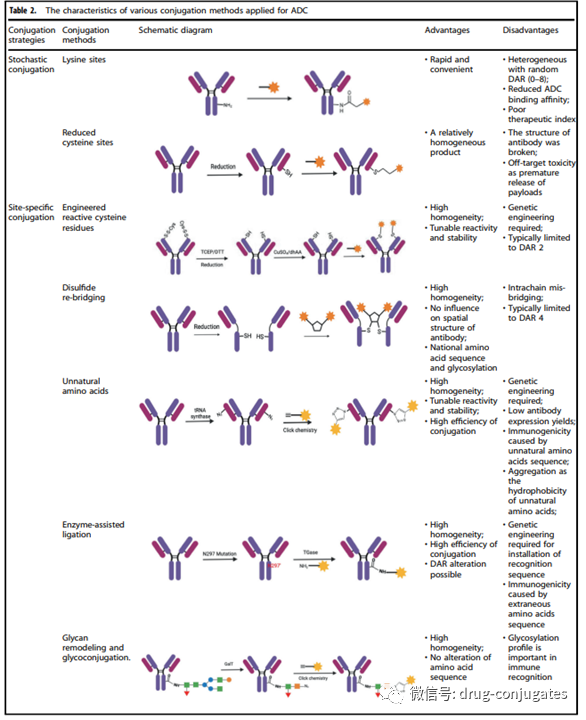
Figure 6 Site-Specific Conjugation Strategies for ADCs (Image Source: Reference [1])
2. Mechanism of Action of ADCs
ADCs exert “specific” targeting effects and “efficient” killing of cancer cells by combining these two mechanisms. These drugs act like precision-guided “biological missiles” that can precisely destroy cancer cells, improve the therapeutic window, and reduce off-target side effects.
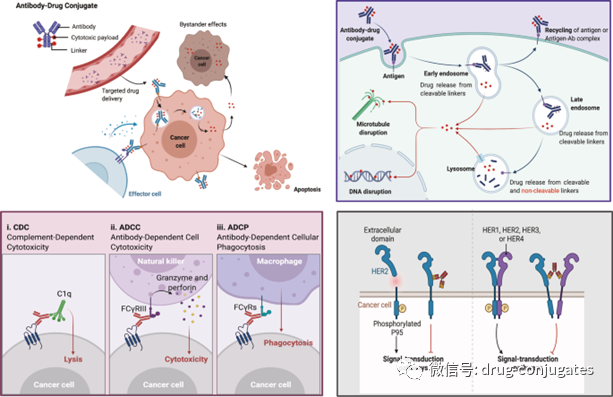
Figure 7 Mechanism of ADC Killing Cancer Cells (Image Source: Reference [1])
The anticancer activity of ADCs also involves the roles of ADCC, ADCP, and CDC. Some Fab fragments of ADC antibodies can bind to antigen epitopes on virus-infected or tumor cells, while the Fc fragment binds to the surface FCR of effector cells (NK cells, macrophages, etc.), thereby mediating direct killing effects (Figure 7 lower left). Additionally, the antibody component of ADCs can specifically bind to the epitopic antigens of cancer cells, inhibiting downstream signal transduction of the antigen receptor (Figure 7 lower right).
3. Research Progress of ADCs
From the perspective of drug composition and technical characteristics, ADC drugs can be subdivided into three generations.
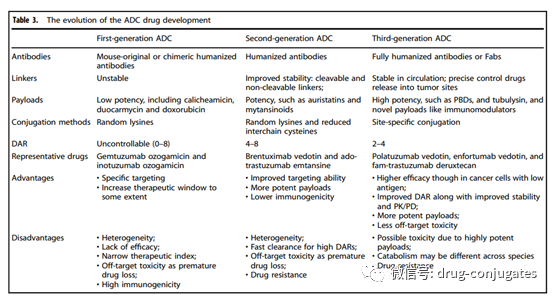
Figure 8 Evolution of ADC Drug Development (Image Source: Reference [1])
First Generation ADCs
Early ADCs were primarily composed of conventional chemotherapy drugs conjugated to mouse-derived antibodies via non-cleavable linkers. The efficacy of these ADCs was not superior to that of free cytotoxic drugs and had significant immunogenicity. Later, the combination of more effective cytotoxic agents with humanized mAbs greatly improved efficacy and safety, leading to the market approval of first-generation ADCs (including gemtuzumab ozogamicin and inotuzumab ozogamicin). In these two products, IgG4 isotype humanized mAbs were used and conjugated to the effective cytotoxic calicheamicin via acid-labile linkers. However, this system also had significant flaws;
1) Instability of the Linker: For example, acidic conditions may arise in other parts of the body, and the linkers in first-generation ADCs can slowly hydrolyze in systemic circulation (pH 7.4, 37°C), leading to uncontrolled release of toxic payloads and unexpected off-target toxicity.
2) Prone to Aggregation: The payloads used in the first generation were hydrophobic and prone to cause antibody aggregation, leading to defects such as short half-lives, rapid clearance, and immunogenicity.
3) Heterogeneity of the Drug: The conjugation of first-generation ADCs was based on random conjugation through lysine and cysteine residues, resulting in a highly heterogeneous mixture with different DARs. Therefore, first-generation ADCs exhibited suboptimal therapeutic windows and required further improvement.
Second Generation ADCs
Represented by Brentuximab vedotin and Ado-trastuzumab emtansine, second-generation ADCs were approved after optimizing mAb isotypes, payloads, and linkers. These two ADCs are characterized by:
1) Use of IgG1 Isotype mAbs, which are more suitable for bioconjugation with small molecule payloads and high tumor cell targeting ability.
2) Higher Toxicity Payloads, improving solubility and conjugation efficiency. More payload molecules can be loaded onto each mAb without inducing antibody aggregation.
3) Improvement of Linkers achieving better plasma stability and uniform DAR distribution. Overall,
improvements in all three elements enhance the clinical efficacy and safety of second-generation ADCs. However, many unmet needs remain, such as insufficient therapeutic windows due to off-target toxicity and aggregation or rapid clearance in ADCs with high DARs. When DAR exceeds 6, ADCs exhibit high hydrophobicity and tend to decrease ADC efficacy due to faster in vivo clearance. In such cases, optimization of DAR through site-specific conjugation, as well as continuous optimization of mAbs, linkers, and payloads, is still needed.
Third Generation ADCs
Third-generation ADCs are represented by polatuzumab vedotin, enfortumab vedotin, fam-trastuzumab deruxtecan, and later approved ADCs.
1) Uniform DAR (2 or 4), with ADCs showing consistent DAR exhibiting less off-target toxicity and better pharmacokinetic efficiency.
2) Fully humanized antibodies instead of chimeric antibodies to reduce immunogenicity.
Additionally, antigen-binding fragments (Fab) are being developed to replace full mAbs in many candidate ADCs, as Fabs are more stable in systemic circulation and may be easier to internalize by cancer cells. Furthermore, more effective payloads have been developed: such as PBD, microtubule inhibitors, and immune modulators with new mechanisms. Although there have been no updates to the types of linkers in the third generation, some new entities for conjugating various payloads have been developed. To avoid interference with the immune system and improve retention time in systemic circulation, more hydrophilic linker combinations, such as PEGylation, have been adopted in third-generation ADCs. Hydrophilic linkers can also be used to balance the high hydrophobicity of certain cytotoxic payloads (like PBD), as ADCs with hydrophobic payloads are generally prone to aggregation. Overall, third-generation ADCs have lower toxicity, higher anticancer activity, and greater stability, allowing patients to receive better anticancer treatments.
4. Current Challenges and Next Generation ADCs
From the approved drugs and candidate drugs in development, it can be seen that the specificity and cytotoxicity of the next generation of ADCs are improving compared to previous generations. However, there are still many challenges in the development of ADCs, including the complexity of pharmacokinetics, inadequate tumor targeting and payload release, and resistance.
Main Challenges
Complex Pharmacokinetic Characteristics
After ADC administration (mainly via intravenous infusion), three main forms may exist in systemic circulation: intact ADCs, naked antibodies, and free payloads. In typical pharmacokinetic characteristics of ADCs, the concentrations of conjugated ADCs and naked antibodies continuously decrease with the internalization of ADCs and antibody clearance. Factors affecting antibody clearance include the mononuclear phagocyte system and Fc receptor (FcRn)-mediated recycling. By binding with internalized ADCs with high affinity, FcRn exports ADCs to extracellular compartments for recycling. Therefore, compared to traditional small molecule drugs, antibodies, including conjugated ADCs and naked antibodies, generally have longer half-lives.
Free cytotoxic payloads are primarily metabolized in the liver and excreted from the body through the kidneys (urine) or feces, which may lead to liver and kidney dysfunction. All these factors, combined with high variability between patients, make it difficult to establish PK and PD models to describe the clinical characteristics of ADCs and assist in designing new ADCs.
Unavoidable Side Effects
Among the 14 approved ADCs, the most common severe side effects (grade 3 or higher) are hematologic toxicities, including neutropenia, thrombocytopenia, leukopenia, and anemia. Hematologic toxicity, as well as liver toxicity and gastrointestinal reactions, may be related to the premature release of cytotoxic payloads into systemic circulation. This is consistent with conventional chemotherapy drugs that primarily affect rapidly proliferating healthy cells. Additionally, the immune responses induced by the antibody component of ADCs may cause secondary damage, leading to renal toxicity. According to recent clinical observations, potential pulmonary toxic effects (such as ILD) during ADC treatment should be noted, especially in anti-HER2 ADCs. In clinical trials of T-DM1 and DS-8201, several deaths were reported associated with ILD. However, the detailed mechanisms of ILD remain unclear. It is speculated that one possible reason may be related to the adverse uptake of ADCs in healthy lung cells and the release of free payloads into the bloodstream. Since the lungs have the richest blood flow and the longest retention time, the adverse uptake of ADCs and free payloads in the blood is most likely to occur in the lungs, inducing ILD. Therefore, appropriate optimizations should be made for the next generation of ADCs to minimize side effects. Adverse reactions should be closely monitored during medication, with preventive or supportive treatment provided.
Tumor Targeting and Payload Release
Compared to traditional cytotoxic drugs, ADCs have a much larger molecular weight, resulting in limited efficiency in penetrating tumors. Current studies indicate that only a small portion of ADCs administered to patients can reach tumor cells, thus the potency of the payload needs to be considered in ADC design.
Resistance
Another challenge in ADC development is resistance. Resistance to tyrosine kinase inhibitors (TKIs) often involves escape mutations in drug targets. However, the resistance mechanisms of ADCs have not been fully characterized. ADC resistance is more complex and diverse. Current evidence suggests that tumors can develop ADC resistance in various ways, such as decreasing antigen expression levels, altering intracellular transport pathways, and developing resistance to the payload.
Next Generation ADCs
1) Targeting Mutated Proteins with ADCs
Current research indicates that the internalization and intracellular transport pathways of ADCs have a critical impact on the cytotoxic activity of ADCs. Compared to wild-type proteins, mutated proteins often have higher levels of ubiquitination, making them easier to internalize and degrade. This means that targeting mutated proteins with ADCs may lead to significant clinical responses. It can be envisioned that ADCs targeting oncogenic mutated proteins (such as certain EGFR mutants) could maximize tumor specificity, reaching levels comparable to selective TKIs.
2) Dual Epitopes or Dual Targeting ADCs
Advancements in bispecific antibody technology have opened up more possibilities for ADC innovation. These ADC designs can improve antibody internalization and enhance tumor specificity. Therapeutics currently under development are exploring these possibilities. Bispecific ADCs targeting different epitopes on the same antigen can improve receptor clustering and lead to rapid internalization of the target. Furthermore, bispecific ADCs targeting both HER2 and LAMP-3 have shown better lysosomal clustering and payload delivery in preclinical studies.
3) Using Two Different Payload Combinations
Using two different cytotoxic agents as payloads in dual payload ADCs can reduce resistance. By accurately controlling the ratio of the two drugs, two synergistic payloads can be delivered to cancer cells, achieving more effective efficacy. Moreover, with the application of two different mechanisms of payloads, the incidence of resistance will be significantly reduced. For example, a homogeneous anti-HER2 ADC containing both MMAE and MMAF was designed, demonstrating significantly better antitumor activity in xenograft mouse models compared to the co-administration of corresponding single payload ADCs.
4) Peptide Drug Conjugates (PDCs)
Another strategy for ADC development is to abandon the traditional structure of mAbs and choose to conjugate the payload with smaller peptide fragments. The main goal of these strategies is to reduce the molecular weight of the ADC, thereby improving penetration efficiency and delivery of the payload to tumor tissues. For example, PEN-221 is an ADC composed of DM-1 conjugated to a peptide chain targeting growth hormone receptor 2. Its molecular weight is only 2 kDa, far below the 150 kDa IgG molecules in traditional ADCs. Currently, such ADCs face technical challenges as they may be rapidly cleared in plasma. However, if we can overcome this barrier, they have the potential to treat hard-to-reach tumors, including poorly vascularized tumors and central nervous system tumors.
5) Developing Non-Internalizing ADCs
Traditionally, to deliver the payload to cancer cells, ADCs require mAbs with high internalization capabilities. However, due to antigen barriers, mAbs often have difficulty diffusing into solid tumor masses. Therefore, non-internalizing antibodies can be developed for ADCs. This is based on the principle of directly releasing the payload into the tumor microenvironment under reducing conditions, which then diffuses into cancer cells leading to cell death.
Finally, there are still many innovative opportunities in the selection of payloads.
5. Conclusion
Currently, various ADC therapies have been successfully developed, benefiting thousands of cancer patients. The approval of 14 ADC drugs and the excellent clinical performance of various ADCs have also attracted more attention to this field, which is very important for this relatively young but highly complex area. With the continuous efforts of researchers in these fields, it is not difficult to imagine that future ADCs will present more surprises in targeted cancer therapy.
References
[1] Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduction and Targeted Therapy (2022) 7:93
[2] The matrix in cancer. Nat Rev Cancer. 2021 Feb 15. doi:10.1038/s41568-020-00329-7. Epub ahead of print. PMID: 33589810.
Original
Optimal Solutions for Improved New Drugs: High-End Formulations
Innovation
Blue Ocean of Innovation: Progress in Domestic Development of Triple and Quadruple Antibodies
Precision
Everything Can Be Conjugated: Overview of Conjugated Drug Technologies and Development Progress
Selection
Next Generation Antibody Drugs: Global Development Progress of Bispecific Antibodies [Collection Edition]
Good
Today’s Star: In-Depth Review of PROTAC Technology and Global Development Progress
Text
The New Drug on the Cutting Edge: R&D Strategies and Case Studies of Class 2.2 (New Dosage Forms)
* This WeChat public account maintains neutrality regarding all original and reprinted content, statements, and opinions. The content is for public sharing only, and if there are any errors in author attribution or copyright issues with reprinted works or images, please kindly remind the original author and contact the editor for deletion.
