–01–
Introduction
Antibody-drug conjugates (ADC) are known as “magic bullets” that precisely target tumor cells through antibodies, delivering cytotoxic drugs while combining the precision of targeted therapy with the potent killing power of chemotherapy. Over the past decade, ADCs have gradually matured in the treatment of solid tumors and hematological malignancies, becoming a revolutionary field in anti-tumor drug research.
In 2009, gemtuzumab ozogamicin (Mylotarg) was the first ADC drug approved by the U.S. Food and Drug Administration (FDA). Currently, the FDA has approved 17 ADC drugs, with hundreds more in clinical research stages.

In 2009, calicheamicin, maytansine, and other drugs were the main cytotoxic agents used for ADC development. Over the past decade, these molecules have continued to be used as payloads for optimization to achieve better stability and hydrophilicity. New cytotoxic agents have also been developed, such as PBDs, duocarmycin, and camptothecin derivatives. Antibody engineering has also made significant progress over the past 10 years, allowing for more site-specific conjugation, improving the homogeneity and stability of ADCs. New second- and third-generation ADCs have entered clinical trials in hopes of achieving better therapeutic effects and safety. Dozens of bioconjugation technologies based on cysteine residues, non-natural amino acids, or molecular engineering models have also been validated in preclinical studies. Furthermore, the identification of more tumor-specific antigen targets and mechanisms for the release of cytotoxic drugs within tumors has led to explosive growth in ADCs, ushering them into a golden age.
–02–
1. First-Generation and Second-Generation ADCs
A successful ADC drug depends on two key factors. First, a stable and reliable linker is needed to connect the antibody and the payload, which remains stable in plasma circulation and is rapidly cleaved after endocytosis by tumor cells to selectively deliver the payload to the tumor while limiting adverse reactions caused by off-target toxicity. The linker must be sensitive to lysosomal conditions (proteases, acidic, and reducing environments).
The second key factor for success is the need to couple a potent cytotoxic agent to the antibody. In fact, due to the lower potency of payloads (e.g., anthracyclines), the first generation of ADCs was characterized by a low therapeutic index, resulting in very limited therapeutic effects even at the maximum tolerated dose (MTD).
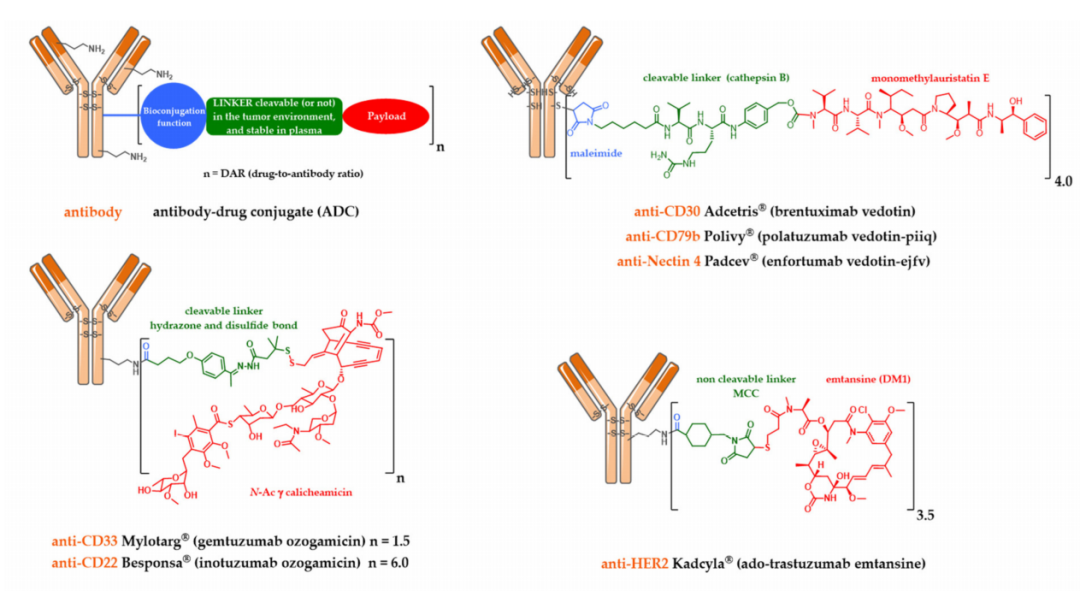
Mylotarg, Besponsa, and First-Generation Cleavable Linkers
Mylotarg was approved by the FDA in 2000 for the treatment of acute myeloid leukemia (AML). It was formed by conjugating calicheamicin to gemtuzumab (a mutated anti-CD33 IgG4 subtype monoclonal antibody) through a cleavable linker containing a hydrazone bond. This ADC had an average drug-to-antibody ratio (DAR) of only 1.5, containing about 50% unconjugated monoclonal antibody. After internalization, the hydrazone bond can be hydrolyzed in the acidic environment of the endosome, releasing the precursor of calicheamicin, which is then reduced by glutathione to free active calicheamicin. The latter binds to the DNA minor groove and undergoes Bergman cyclization, generating highly reactive double radicals that cause sequence-selective DNA double-strand breaks.
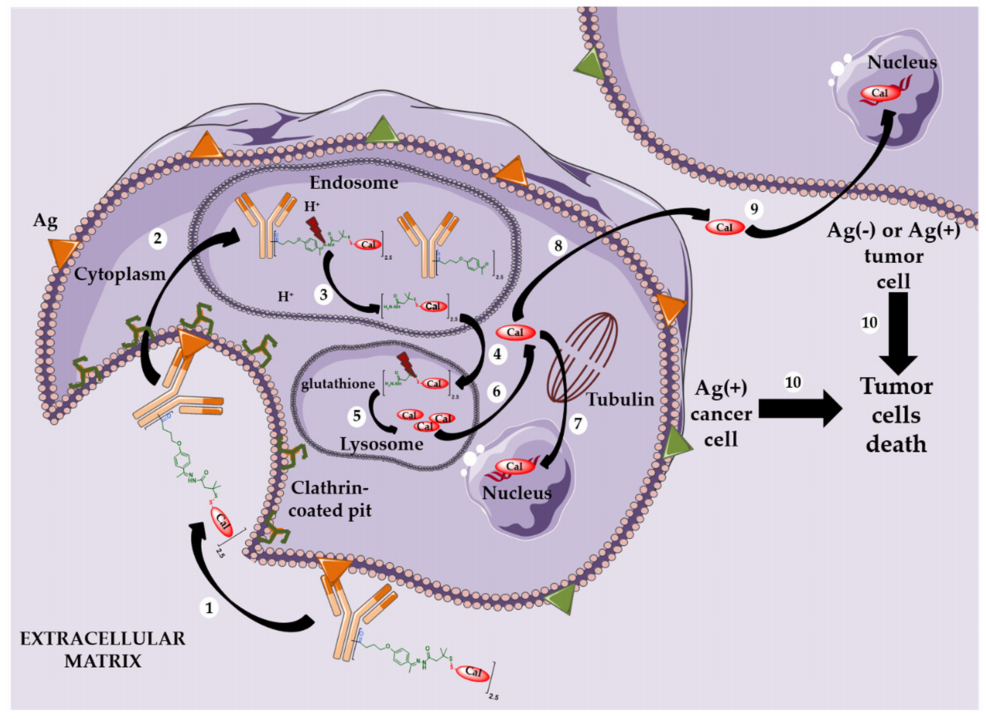
Theoretically, the hydrazone bond should remain stable in blood circulation at physiological pH and undergo selective hydrolysis after internalization under acidic conditions. However, the linker of Mylotarg exhibited some instability, leading to premature release of calicheamicin in plasma circulation, resulting in severe toxicity that led Pfizer to withdraw Mylotarg from the market in 2010.
Thanks to the clinical experience accumulated in recent years and technological advancements, Mylotarg was re-approved in 2017 with optimized linker stability, used at lower doses, and modified dosing schedules suitable for different patient populations.
A similar linker was developed and used to conjugate calicheamicin to inotuzumab, a mutated CD22-targeting antibody, which was approved by the FDA in 2017 for the treatment of acute lymphoblastic leukemia (ALL).
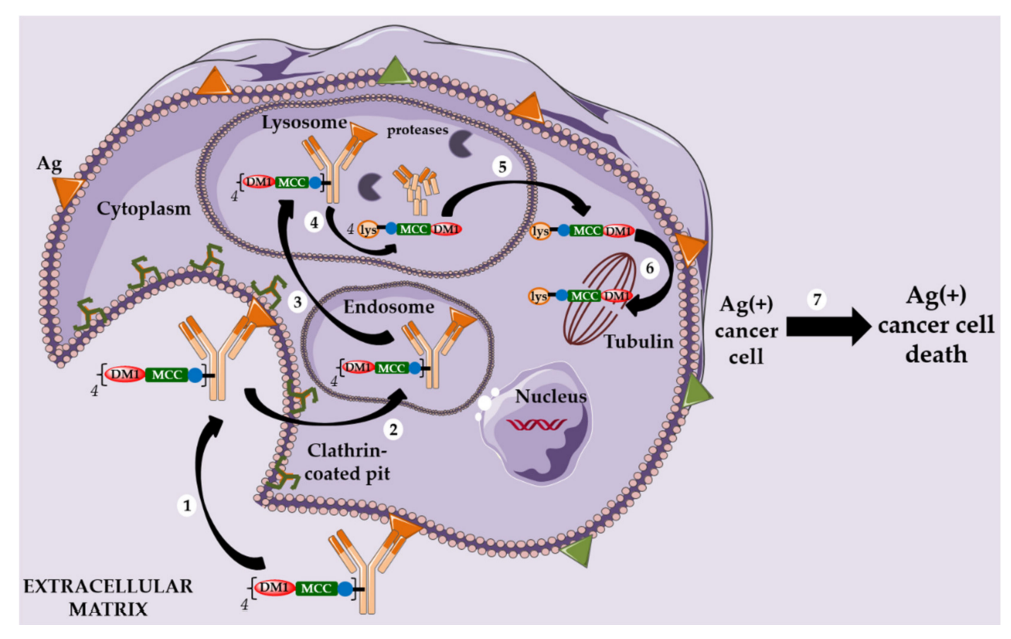
Kadcyla and Second-Generation Non-Cleavable Linkers
In light of these findings, alternative strategies for linker design continued to be developed. However, a serendipitous discovery allowed Immunogen to identify an unexpectedly effective ADC. DM1 was conjugated to the lysine residues of trastuzumab through a non-cleavable linker containing N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC), and this ADC (T-DM1, Kadcyla) was approved by the FDA in 2013 for HER2-positive breast cancer patients.
This novel ADC was highly effective in HER2-positive breast cancer models, with the original structure only becoming active after complete digestion by enzymes in the lysosome following internalization, yielding the active metabolite Lys-MCC-DM1.
Adcetris, Polivy, and Second-Generation Cleavable Linkers
Meanwhile, Seattle Genetics designed its own conjugation technology using a cleavable linker mc-VC-PABC, which includes a maleimide-based spacer, a standard Val-Cit dipeptide sequence as a substrate for cathepsin, and a self-degradable spacer, to bioconjugate monomethyl auristatin E (MMAE) to the cysteine residues of anti-CD30 antibodies. This ADC (Adcetris) was approved by the FDA in 2011 for the treatment of anaplastic large cell lymphoma and Hodgkin lymphoma.
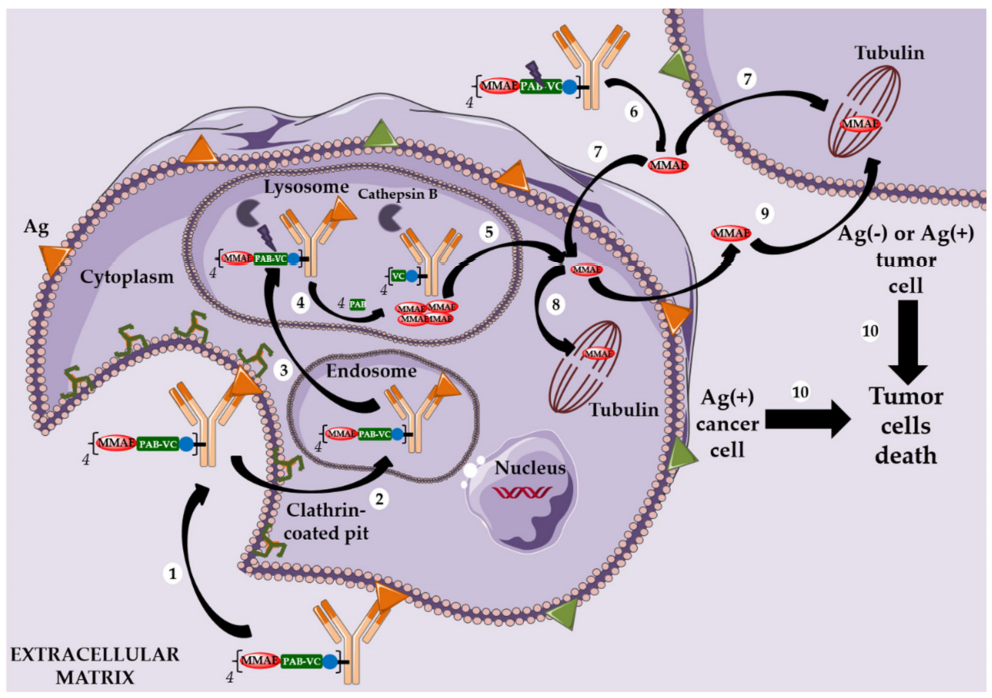
After internalization in tumor cells, the cleavable linker of Adcetris degrades, releasing MMAE, which can destroy target cells and spread to the cell membrane, reaching and killing adjacent cancer cells. This phenomenon is known as the bystander effect, allowing released MMAE to kill both CD30-positive and CD30-negative tumor cells.
Similarly, this second-generation linker (mc-VC-PABC) was used in Polivy, an ADC that conjugates MMAE with polatuzumab (anti-CD79b monoclonal antibody), which was approved by the FDA in June 2019 for the treatment of adult diffuse large B-cell lymphoma (DLBCL).
–03–
2. Third-Generation ADCs
Due to the interference related to the internalization, transport, or recycling of monoclonal antibodies in the first two generations of ADCs, the shedding of antigens, and the lysosomal degradation defects of ADCs, there is a need to develop new technologies related to bioconjugation, carrier forms, linkers, or toxic drugs to broaden the application of ADCs, leading to the emergence of third-generation ADCs.
Site-specific ADCs
Despite the increasing success of ADCs, until 2019, every approved ADC on the market existed as a heterogeneous mixture, with varying numbers of cytotoxic drugs on the monoclonal antibodies distributed in different locations, leading to analytical issues during production. In fact, the DAR is uncontrolled, and this complex mixture significantly affects the pharmacokinetics and efficacy of ADCs.
Unconjugated antibodies may act as competitive inhibitors, and weak DAR conjugates exhibit poor efficacy, while antibodies with high DAR are rapidly eliminated from plasma, impairing the therapeutic window of ADCs. To broaden the therapeutic index of ADCs, region-specific bioconjugation methods have been widely developed since 2008. These site-specific methods can be divided into three categories: (i) bioconjugation using natural or non-natural amino acids, (ii) enzyme-mediated bioconjugation, or (iii) linker-based bioconjugation.
The first method involves introducing specific amino acids through antibody engineering. Junutula and his colleagues at Genentech were pioneers in developing an ADC by site-specific bioconjugation of a monoclonal antibody targeting the ovarian cancer antigen MUC16 with the Adcetris® linker at the 265th amino acid. To achieve this, they introduced two cysteine residues into the amino acid sequence of the monoclonal antibody, selecting these two cysteines to maintain IgG folding and binding to the antigen.
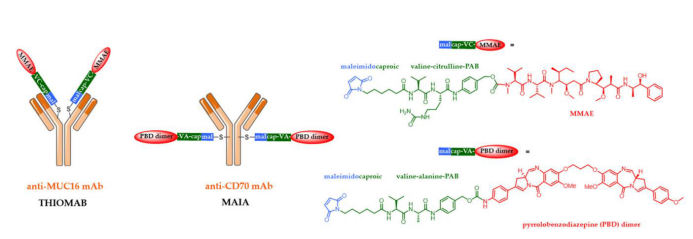
The resulting TDC (ADC-thioantibody) was compared with ADCs generated using traditional random bioconjugation methods.
Both ADCs were effective in mouse xenograft models, but TDC exhibited higher tolerance in rats and cynomolgus monkeys and demonstrated lower systemic toxicity in vivo. Inspired by this strategy, Seattle Genetics and Spirogen developed a similar technology called MAIA, achieving bioconjugation of PBD dimers by introducing a serine-cysteine mutation at the 239th position of the mAb critical region.
The second possibility is to use enzyme-mediated region-specific bioconjugation technology. Transglutaminase catalyzes the formation of amide bonds between the glutamine side chains of natural monoclonal antibodies and molecules containing primary amines. Based on this, Innate Pharma has developed a three-step method for constructing ADCs using transglutaminase, as shown in the figure below.
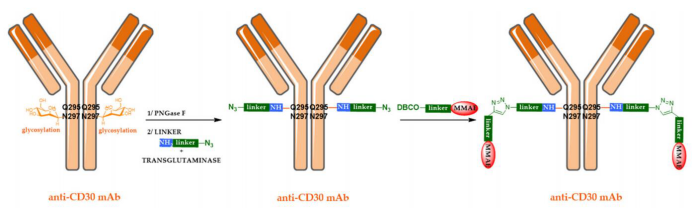
Finally, region-specific ADCs can also be derived from natural monoclonal antibodies.
In this strategy, heterogeneous bifunctional linkers composed of innovative dibromomaleimide (DBM) or dithiophenyl maleimide (DSPh) can produce more uniform and stable ADCs through region-specific bioconjugation, achieving a DAR of 4.
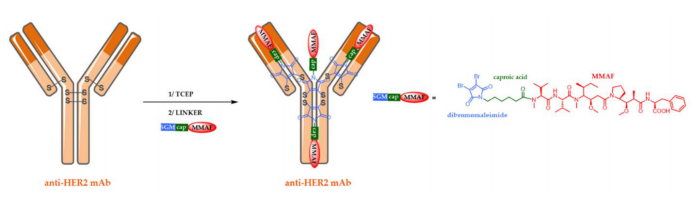 Other Forms of ADCs
Other Forms of ADCs
Despite their significant efficacy, most ADCs targeting solid tumors have not progressed beyond phase II clinical trials, indicating that further optimization of other parameters is needed to enter the market. The effectiveness of ADCs is limited by their size (150 kDa) and poor penetration and absorption in tumors. Besides the size of IgG, it is now believed that the Fc portion of IgG is unnecessary for the efficacy of ADCs and may even be detrimental; in fact, the longer half-life of ADCs induced by FcRn increases exposure to healthy tissues, while FcγR cross-reacts with endothelial cells and the immune system, both of which are associated with off-target toxicity.
Smaller conjugation forms have been explored to address these shortcomings, particularly peptides, single-domain antibody fragments (sdAb or VHH), single-chain antibodies (scFv), antigen-binding fragments (Fab), or small immunoproteins (SIP) that dimerize using the CHε4 domain. As part of this strategy, recently, auristatin derivatives were site-specifically conjugated to anti-HER2 single-chain antibodies (derived from trastuzumab) to generate two new single-chain antibody-drug conjugates (SDCs).
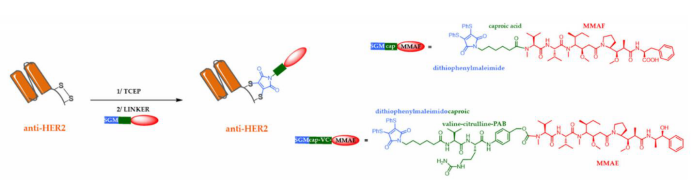
Compared to natural single-chain antibodies, these two SDCs maintained affinity for HER2 and effectively killed SK-BR-3 HER2-positive cells in vitro (EC50 values of 0.68 nm and 0.32 nm), with no effect on HER2-negative MCF-7 cells.
Although SDCs with a DAR of 1 are not as potent as corresponding ADCs with a DAR of 4, this work represents the first step towards designing more effective small conjugates with higher DARs, paving the way for further in vivo or even clinical studies to assess their potential against solid tumors.
Currently, ADCs targeting HER2 are ineffective in clearing HER2+ cancer cells with relatively low expression. Therefore, only about 20% of breast cancer patients are eligible for HER2-targeted therapy. Additionally, the intratumoral heterogeneity of HER2 expression ultimately leads to relapse in patients who initially respond to treatment. To achieve effective anti-tumor activity in cancer cells with a broader range of HER2 expression, MedImmune has developed a dual-wing ADC, IMMU4276, targeting HER2.
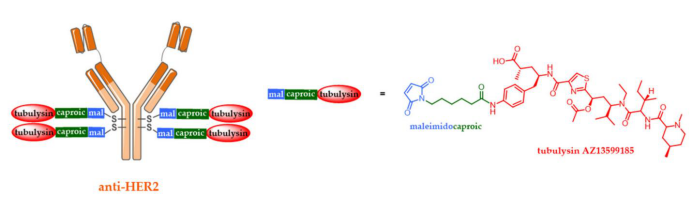
Compared to Kadcyla®, this dual-wing ADC demonstrated superior anti-tumor activity in various tumor models representing different patient subgroups. Furthermore, two combination mutations (L234F and S239C) reduced binding to FcγR to decrease FcγR-mediated internalization of ADCs in normal tissues, thereby reducing the occurrence of side effects such as thrombocytopenia. Unfortunately, due to high hepatotoxicity, this ADC was also halted in clinical trials in 2018.
New Targets and Related Release Systems
In solid tumors rich in interstitial stroma, targeting internalizing antigens on cancer cell surfaces is very challenging. Therefore, a new approach has been developed that targets the tumor microenvironment (stroma or vascular system) instead of cancer cells. In this strategy, extracellular proteases and other components of the extracellular matrix (such as acidic media or reducing glutathione) can be used for effective extracellular release of cytotoxic agents for non-internalizing ADCs.
Fibronectin is a component of the extracellular matrix beneath tumor endothelium, which is more accessible for immune binding compared to cancer cell surfaces. The SIP form results from the fusion of a single-chain antibody fragment with the human IgEεCH4 domain. SIP (F8) is generated at the C-terminus with two unpaired cysteine residues, allowing two DM1 molecules to undergo region-specific bioconjugation to produce SIP (F8)-SS-DM1 with a DAR of 2.
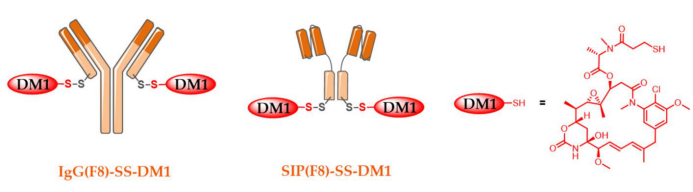
In therapeutic experiments, SIP (F8)-SS-DM1 outperformed its IgG (F8)-SS-DM1 counterpart.
In the F9 teratoma mouse model, a comparison of the two conjugates was made in vivo with equal (stoichiometric) doses administered in five injections of 5 mg/kg. The SIP conjugate achieved complete tumor remission in four-fifths of the mice, while limited tumor remission was observed with the IgG conjugate. Significant differences were also noted in the release kinetics of cytotoxic DM1 between the two conjugates. The advantage of non-internalizing ADCs is that they bypass certain resistance mechanisms associated with immune conjugate internalization, while region-specific bioconjugation methods control DAR, avoiding the emergence of components with a DAR of 0.
New Cytotoxic Drugs
New cytotoxic drugs have been developed to target cancer cells with low antigen expression or those resistant to auristatins or maytansinoids. To this end, PBD dimers have been developed, which contain two alkylating imine functional groups capable of forming covalent bonds with DNA. PBD dimers are approximately 50-100 times more potent than traditional cytotoxic drugs (MMAE or DM1). Spirogen and Seattle Genetics introduced PBD dimers as cytotoxic agents in ADCs and developed SGN-CD33A (vadastuximab-talinine) and SGN-CD70A; however, unfortunately, both PBD-based ADCs were recently forced to halt in phase III clinical trials. SGN-CD33A was associated with fatal infections and a higher patient mortality rate due to potential hepatotoxicity, while SGN-CD70A was associated with thrombocytopenia.
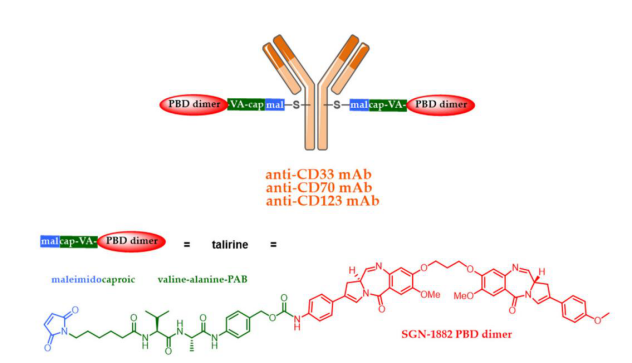
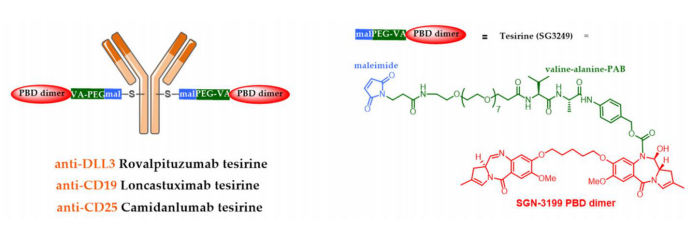
Although the use of talirine ADCs was halted during clinical trials, two ADCs using the corresponding tesirine are in advanced stages of clinical research. Rovalpituzumab tesirine (Rova-T or SC16LD6.5) is an anti-DLL3 ADC developed by AbbVie (Stemcentrx) that has been tested in phase III clinical trials for small cell lung cancer.
ADC Therapeutics has developed loncastuximab-tesirine (ADCT-402) and camidanlumab-tesirine (ADCT-301), both of which are being tested in critical phase II clinical trials for B-cell acute lymphoblastic leukemia and Hodgkin lymphoma, respectively.
Genentech has developed a new anti-CD22-NMS249 ADC, with a new anthracycline compound named PNU-159682. Anthracyclines are among the most widely used chemotherapeutic agents and are very effective in treating aggressive non-Hodgkin lymphoma (NHL). This ADC uses an MC-VC-PAB-DEA linker with an extended self-locking spacer, which includes N,N’-dimethyl-ethylenediamine (DEA). In vivo, this anti-CD22-NMS249 ADC was as effective in xenograft models as the anti-CD22-VC-MMAE ADC, and it remained effective in cell line models based on the anti-CD22-VC-MMAE ADC, demonstrating the efficacy of anthracycline ADCs in treating MMAE-resistant cancers.
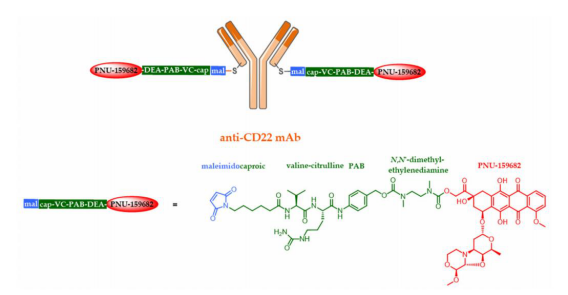
As an alternative to the previously mentioned ADCs, Heidelberg Pharma has developed a patented technology called ATAC (Antibody Targeted Amanitin Conjugates) based on strong RNA polymerase II inhibitors. Alpha- and beta-amanitin are two well-known amanitin toxins identified in mushrooms over forty years ago. HDP-101 is generated by site-specific conjugation of HDP30.2115 (stable analog of alpha-amanitin) with a protease B-sensitive linker targeting B-cell maturation antigen (BCMA, CD269) designed by Genentech, resulting in an ATAC with a DAR of 2.
BCMA has become a highly selective target for treating multiple myeloma. In vivo, HDP-101 led to complete tumor remission in mice across various xenograft models of multiple myeloma, and safety studies in mice and monkeys identified a very favorable therapeutic window. ATACs represent a very promising class of compounds, but they are still in the preclinical stage.
The Success Stories of Third Generation: Enhertu and Trodelvy
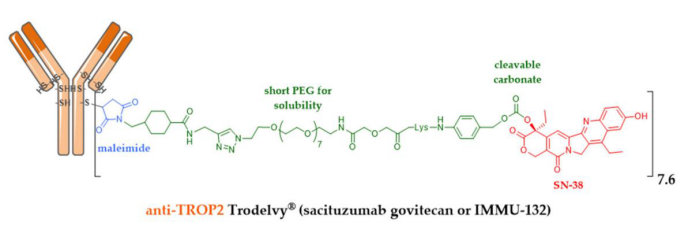
Among many companies developing ADCs, Immunomedics designed a surprising ADC through a triple bet: constructing an ADC targeting a slightly overexpressed target, using a system that mixes intracellular and extracellular release, and employing a less potent cytotoxic drug than conventionally used. Sacituzumab-govitecan (IMMU-132) is an anti-TROP-2 monoclonal antibody conjugated to SN-38 (the active metabolite of irinotecan) through a cleavable maleimide linker with short polyethylene glycol units.
Approved by the FDA in April 2020, this achievement is impressive as this ADC is used for difficult-to-treat or resistant triple-negative breast cancer (TNBC), for which there were no effective therapeutic agents previously. Another interesting feature of this ADC is that the optimization of the linker structure, which includes polyethylene glycol units, allows for a DAR of up to 7.6 without affecting its tolerability or efficacy. A DAR of 4 has long been considered optimal, but this claim now only applies to known recognized ADCs with second-generation linkers whose effective payloads are DM1 or MMAE.
Similarly, to enable the conjugation of irinotecan derivatives with carefully designed linkers, Daiichi Sankyo developed DXd (exatecan or DX-8951). DXd is a cytotoxic agent that is 10 times more active against cancer cells than SN-38 in vitro. DXd has better safety and optimal solubility, capable of inducing bystander killing effects to kill adjacent cancer cells, which is an advantage in heterogeneous tumors, but has a short half-life to avoid off-target toxicity.
By bioconjugating DXd to the cysteine residues of anti-HER2 trastuzumab through a proteolysis-sensitive maleimide linker, a conjugate with a uniform DAR of 7.7 was obtained, fam-trastuzumab-deruxtecan nxki (DS-8201a). Despite the high DAR, DS-8201a from Daiichi Sankyo demonstrated excellent tolerability in rats and monkeys and was very stable in plasma.
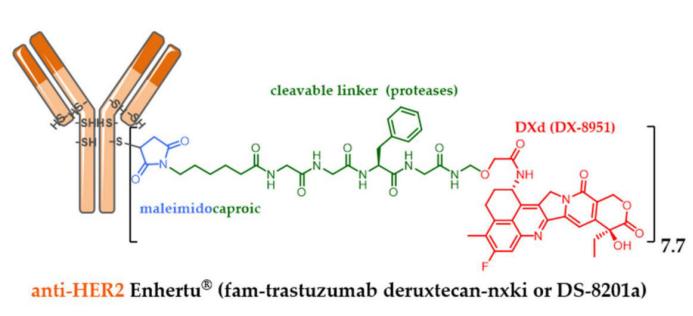
DS-8201a effectively delivers DXd to heterogeneous tumors in vivo and shows high therapeutic efficacy. Last year, in a phase III clinical study of metastatic HER2-positive breast cancer, DS-8201a successfully compared with T-DM1 and was ultimately approved by the FDA at the end of December 2019.
–04–
3. Toxicity of ADCs
Gemtuzumab-Ozogamicin
Gemtuzumab ozogamicin (Mylotarg) was approved by the FDA in 2000 for the treatment of certain patients with acute myeloid leukemia (AML). In 2001, the FDA issued a warning due to observed cases of venous occlusive disease. In 2004, a randomized study comparing traditional therapy with Mylotarg was prematurely stopped due to increased mortality associated with the latter. Mylotarg was withdrawn from most markets, except Japan, in 2010.
Due to limited analytical methods available during the first clinical studies, Mylotarg was later identified as a heterogeneous product in terms of DAR. Although the theoretical DAR value is around 2.5, over 50% of the antibodies in the drug were unconjugated, while other antibodies had DAR values of 4 or 5. At the same time, Mylotarg’s determination of a single dosing regimen made it difficult to achieve a satisfactory therapeutic index, leading to the abandonment of its development.
However, subsequent studies by the French Acute Leukemia Association (ALFA) indicated that dividing the dosing into three doses improved survival rates without serious adverse events. Thus, Mylotarg was re-approved for marketing in 2017.
Brentuximab Vedotin
Brentuximab vedotin (Adcetris) was approved in 2011 for the treatment of certain CD30-expressing lymphomas, including Hodgkin lymphoma and anaplastic large cell lymphoma. However, Adcetris as a monotherapy exhibited potential severe peripheral neuropathy, neutropenia, and thrombocytopenia, which are typical side effects of anti-platelet drugs. On the other hand, rare but severe cases of progressive multifocal leukoencephalopathy were observed.
Additionally, the combination of Adcetris with bleomycin (a commonly used drug for treating Hodgkin disease) was found to lead to unacceptable pulmonary toxicity, and this combination was therefore excluded.
Typical or Unexpected Toxicities of ADCs
Based on the classification of ADC payload drugs, such as microtubule disruptors (auristatins and maytansinoids), anti-mitotic inhibitors (KSPis), or DNA-damaging drugs (calicheamicin and PBD), certain toxicities can be expected. These include bone marrow toxicity that can reach grade 4, sensitive nerve and growth toxicities.
On the other hand, some side effects that are not observed with standard cytotoxic drugs have also been reported. These include ocular toxicities, such as keratitis or corneal deposits caused by ADCs containing MMAF or DM4, which may constitute limiting toxicities for these ADC drugs. Kadcyla has been shown to increase the risk of radiation necrosis. Understanding and managing these unexpected toxicities is crucial for optimizing the use of these drugs.
–05–
4. Mechanisms of ADC Resistance
ADCs act on tumor target cells through several stages: binding to antigens, internalization, drug release (primarily in lysosomes), drug release into the cytoplasm, and drug action on targets inducing apoptosis. Each of these steps may be associated with resistance suggested by preclinical studies in vitro or in vivo:
(i) Downregulation of target antigens and/or defects in antibody binding, internalization, transport, or recycling;
(ii) Defects in lysosomal degradation of ADCs or reduced expression of lysosomal transporters such as SLC46A3, leading to decreased release of payloads into the cytoplasm;
(iii) Alterations in microtubule proteins or microtubule dynamics regulators;
(iv) Reduced drug retention in cells through upregulation of resistance transporters such as MDR1.
The clinical relevance of these potential resistance mechanisms remains to be confirmed. In fact, obtaining tumor samples before ADC treatment and during relapse after treatment is very complex. Additionally, in the context of combination therapies, distinguishing resistance mechanisms to ADCs from other drugs is equally complex.
–
06
Conclusion
Over the past decade, ADCs have improved through the selection of better cytotoxic drugs, bioconjugation methods, better-targeted antigens, and optimized antibody engineering. However, they still face some limitations (such as limited penetration in solid tumors and toxicity) and the emergence of resistance mechanisms.
To overcome these limitations, new antibody forms, new delivery systems, non-internalizing antigen targets, new cytotoxic drugs, and site-specific bioconjugation methods have been explored to promote the development of ADCs. Although many innovations have yet to be validated in clinical protocols, research in this field has provided us with many encouraging results. It is believed that the next decade for ADCs will usher in a more brilliant future.
References:
1. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245
Source: 小药说药 2025-08-11