Introduction
Degrader-antibody conjugates (DAC) are a novel entity that combines the payload of a proteolysis-targeting chimera (PROTAC) with monoclonal antibodies through a type of chemical linker (DAC = PROTAC + ADC). This article collects several examples of DAC from scientific and patent literature and records specific challenges associated with DAC construction. Overall, these examples indicate that DACs with bioactivity can be successfully prepared using various PROTAC payloads.
Author | Glucose
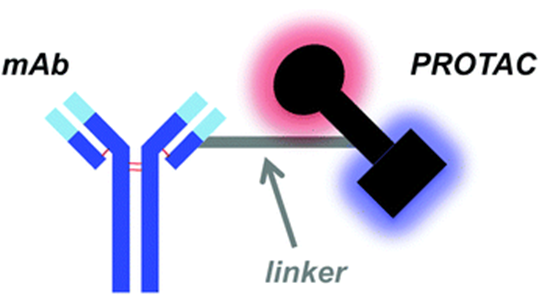
Figure Caption: Schematic structure of DAC
Some Key Points of DAC
(1) DAC consists of mAb, linker, and PROTAC payload.
(2) Match the expression of target antigens with the degradation payload cell biology.
(3) The PROTAC payload must be stable in lysosomes and able to escape safely.
(4) A DAR >4 may be required, which could affect conjugation and DAC pharmacokinetics.

Introduction to PROTAC
Before discussing DAC, we need to understand what PROTAC is. Have you heard of this trending technology?
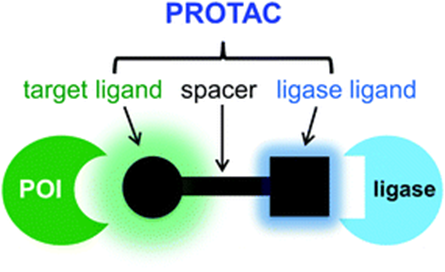
PROTAC, or “proteolysis-targeting chimera,” is rapidly transforming the fields of biology and medicinal chemistry. This bifunctional molecule can specifically degrade intracellular target proteins, thus holding potential for improved and/or prolonged bioactivity compared to simple small molecule inhibitors of the same entity.
The structure of PROTAC is shown in the figure below: it typically consists of a recognition element for the target protein, a portion that binds to E3 ligase, and a spacer group that connects these two components (For specific mechanisms, please refer to “ubiquitination degradation”).
However, PROTACs have poor DMPK properties, such as low oral bioavailability and/or rapid in vivo clearance. This has become a significant barrier to the development of PROTACs, along with issues such as large molecular weight (700-1100Da) and limited available E3 ligases.

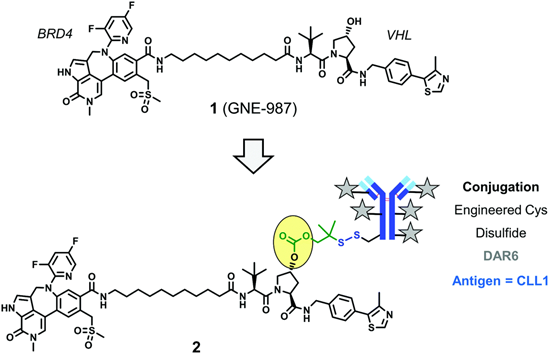
A DAC Targeting BRD4
In early 2020, Genentech described a highly efficient DAC conjugate based on VHL for degrading bromodomain-containing protein 4 (BRD4) (GNE-987), linked between ADC and PROTAC via a cleavable disulfide bond, targeting the protein CLL1 (C-type lectin-like molecule-1), with a DAR of 6.
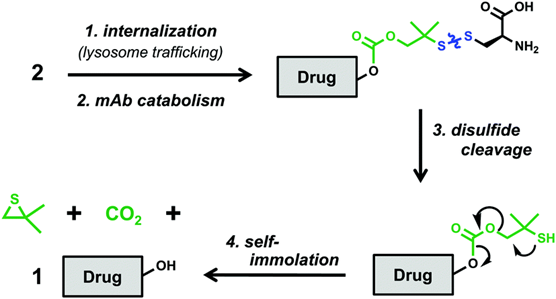
The mechanism of action of this DAC may be:
The antibody part of the DAC binds to the CLL1 receptor on the surface of tumor cells, after which the conjugate is internalized and transported to lysosomes (Step 1). In this proteolytic environment, the antibody is metabolically broken down into amino acid components, leaving a cysteine residue that is partially attached to the remainder of the linker via a disulfide bond (Step 2). The disulfide bond is then reduced (Step 3) to provide the corresponding thiol, which undergoes self-immolation to release the degrader 1 to exert its effect (Step 4).
In the HL-60 (left) and EOL-1 (right) xenograft models, a single intravenous dose showed that:
(1) Unconjugated CLL1 mAb had no significant activity in these models. (2) The CLL1 degrader conjugate with GNE-987 hydroxyproline isomer payload also showed no activity. (3) The correctly structured DAC targeting CLL1 exhibited dose-dependent activity, with significant tumor suppression effects. (4) Unconjugated GNE-987 had no activity.
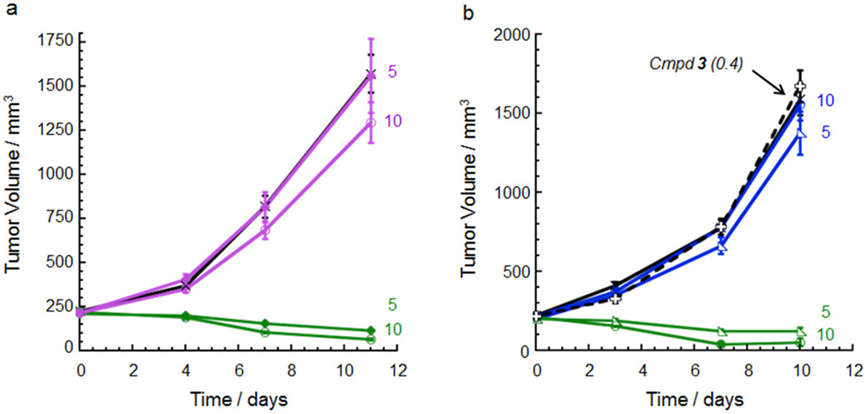
Figure Caption: Green trace: Degrader-mAb conjugate ; Purple trace: Compound isomer DAC
Blue trace: mAb Black trace (dashed): Unconjugated compound

Another DAC Targeting BRD4
Shortly after the above DAC appeared, University College London published a second example of a BRD4 degrader-antibody conjugate. They used dibromomaleimide reactions to reduce interchain disulfide bonds of mAb, generating cysteine residues for connecting four copies of relevant linkers-drugs via a thiol rebridging method.
They used an ester moiety (yellow oval) to connect the VHL-based PROTAC payload to an uncleavable linker, and completed the final linker structure cycloaddition (SPAAC) reaction using copper-free, strain-promoted azide-alkyne cycloaddition. After delivering the conjugate to lysosomes, intracellular payload release can occur via ester cleavage. The antibody targets HER2.
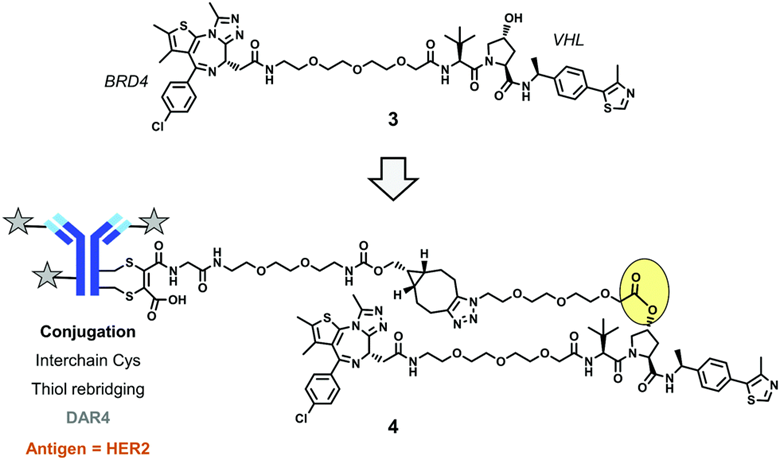
In vitro experiments showed that this DAC provided dose-dependent BRD4 degradation in two HER2-positive cell examples but none in two HER2-negative control cell lines.
Moreover, fluorescence labeling observed DAC internalization in HER2-positive cells, followed by transport to lysosomal compartments. No related internalization and transport were detected in HER2-negative cells, consistent with the lack of degradation activity observed in experiments using these cell lines.

Future Outlook
1. Although the field of degrader-antibody conjugates DAC is still in its infancy, various such entities have already been created, with validated DACs showing meaningful in vitro and/or in vivo bioactivity.
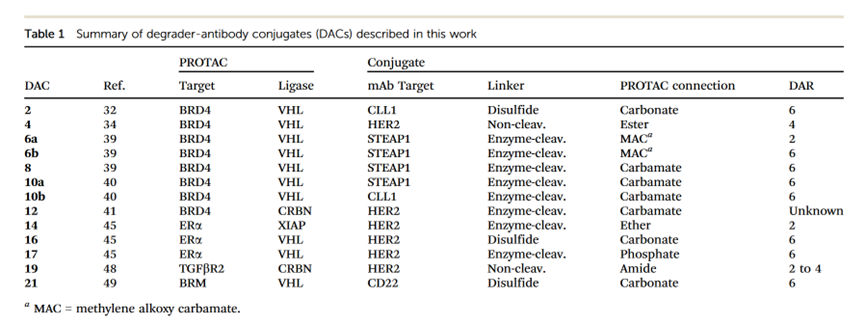
2. In the generation of these conjugates, PROTACs binding to several different E3 ligases were used, which degraded several well-differentiated target proteins (Table 1). Additionally, many different novel linkers and mAb conjugation methods were employed to construct the described DACs (Table 1).
3. Compared with most known cytotoxic ADCs (DAR = 2 to 4), many of these new entities adopted higher payload loads (DAR values of 6), but whether this increase is typically necessary for DAC applications remains to be determined.
4. Challenges still exist in determining which PROTACs are suitable for DAC conjugation and how best to maintain (and ideally enhance) the bioactivity of the selected chimera degrader.
References:
[1] Dragovich PS. Degrader-antibody conjugates. Chem Soc Rev. 2022 May 23;51(10):3886-3897. doi: 10.1039/d2cs00141a. PMID: 35506708.
[2] Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. ChemMedChem. 2020 Jan 7;15(1):17-25. doi: 10.1002/cmdc.201900497.
[3] M. Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306.

“Precision Medicine” public account created by Dr. Zhang Zhang from Jinan University School of Pharmacy.Dr. Zhang Zhang is mainly engaged in: 1) Discovery, mechanism, and druggability research of small molecule targeted anti-tumor drugs; 2) Research on disease and drug target discovery based on chemical proteomics. As a principal investigator, the team has developed 4 kinase inhibitors that have been successively transferred to companies for later development, among which 1 has been marketed (Orelabrutinib), 1 is in production, 1 is in Phase I clinical trials, and 1 is in preclinical research stage.
The laboratory has a complete technical platform for in vivo and in vitro efficacy, mechanism, druggability, target discovery, and validation; supporting and promoting the discovery, druggability evaluation, new indications, and mechanisms of various enterprise-targeted anti-tumor drugs.Dr. Zhang Zhang is committed to promoting the research and development of original new anti-tumor drugs and new targets, widely collaborating with research institutes and corporate teams. For more information and consulting cooperation:

Disclaimer: The publication/reproduction of this article is merely for the purpose of disseminating information and does not imply endorsement of or verification of its content by this public account. Any judgments made based on this content are at your own risk.If there is any infringement, please notify for deletion!
Long press to follow this public account
 Fan Group/Submission/Authorization/Advertisement etc.Please contact the public account assistant
Fan Group/Submission/Authorization/Advertisement etc.Please contact the public account assistant  If you find this article interesting, please click here↓
If you find this article interesting, please click here↓