Source: Beike Society
Antibody-drug conjugates (ADCs) have become a popular avenue in tumor treatment in recent years, typically covalently linking a monoclonal antibody (mAb) to a cytotoxic drug (payload) via a chemically synthesized linker. ADCs combine the advantages of highly specific targeting capabilities with potent killing effects, achieving precise and efficient elimination of cancer cells.
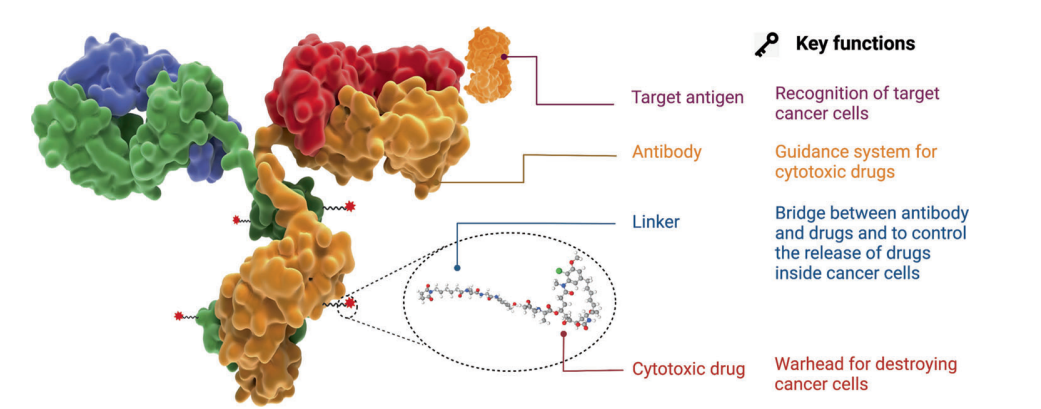
Figure 1. Structure and Characteristics of ADC Drugs
Since the FDA approved the first ADC product, Mylotarg® (gemtuzumab ozogamicin), in 2000, by December 2021, a total of 14 ADCs have received market approval worldwide for hematological malignancies and solid tumors. Additionally, there are over 100 ADC candidates currently in various stages of clinical trials.
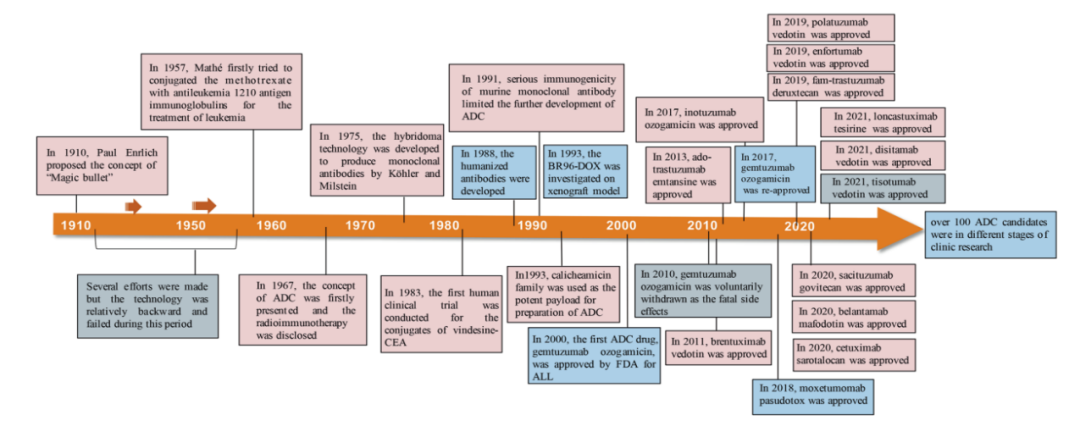
Figure 2. Key Events in the Development and Approval Process of ADC Drugs
However, the path to developing innovative tumor products is not always smooth, and the ADC field is no exception. For example, the first globally marketed ADC product, Mylotarg®, and the recently failed Phase III clinical trial of Blenrep®.
Failure of Blenrep® Phase III Study
-
On November 7, 2022, GSK’s targeted BCMA ADC product Blenrep® (belantamab mafodotin) failed in a Phase III clinical trial (DREAMM-3) involving patients with a type of blood cancer, as it did not meet the primary endpoint of progression-free survival (PFS) compared to the control group receiving monotherapy with pomalidomide and low-dose dexamethasone. Previously, Blenrep (belantamab mafodotin) had received accelerated approval from the FDA as a monotherapy for adult patients with relapsed or refractory multiple myeloma (RRMM) who had received at least four prior treatment regimens.
Two Launches of Mylotarg®
-
In 2000, the FDA granted accelerated approval to Pfizer’s CD33-targeted ADC product Mylotarg® as a monotherapy (high dose) for adult patients with CD33-positive AML who had experienced their first relapse, were ≥60 years old, and were unsuitable for other cytotoxic chemotherapy. This marked Mylotarg®’s first launch, making it the world’s first commercialized ADC product. However, shortly after its launch, due to safety concerns, Pfizer announced the withdrawal of Mylotarg® from the U.S. market in 2010.
-
On September 1, 2017, Pfizer received FDA approval again by updating clinical evidence, adjusting drug specifications, and dosage regimens: (1) for newly diagnosed CD33-positive AML adult patients; (2) for children and adult patients aged ≥2 years with CD33-positive, relapsed or refractory AML.
The efficacy and safety of ADCs, therapeutic windows, target limitations, and the heterogeneity of solid tumors and hematological malignancies all pose challenges to ADC development.
This article aims to review the development history and mechanisms of ADCs, explore key components of ADCs and how these key factors affect ADC activity, and predict the future development prospects of next-generation ADC products.
Selection of Antigens for ADCs
The target antigens expressed on tumor cells guide ADC drugs in recognizing tumor cells and determine the mechanism of delivering cytotoxic payloads into tumor cells (e.g., endocytosis). Therefore, the appropriate selection of target antigens is the primary consideration in ADC development.
-
First, to reduce off-target toxicity, the targeted antigens should be expressed or predominantly expressed in tumor cells but minimally or not at all in normal tissues. Ideally, the antigens should be surface (or extracellular) antigens rather than intracellular antigens so that circulating ADCs can recognize them. For example, the expression of HER2 receptors in certain types of tumors is about 100 times higher than in normal cells, laying a solid foundation for the development of ado-trastuzumab emtansine (TDM-1, KADCYLA®), fam-trastuzumab deruxtecan (DS-8201a, Enhertu®), and disitamab vedotin (RC48, Aidixi®);
-
Second, the target antigens should ideally be non-secreted, as secreted antigens in circulation can lead to undesirable ADC binding outside the tumor site, resulting in reduced tumor targeting and increased side effects;
-
Third, the target antigens should ideally be internalized upon binding with the corresponding antibody, allowing the ADC-antigen complex to effectively enter cancer cells, followed by appropriate intracellular transport pathways and rapid release of the cytotoxic payload.
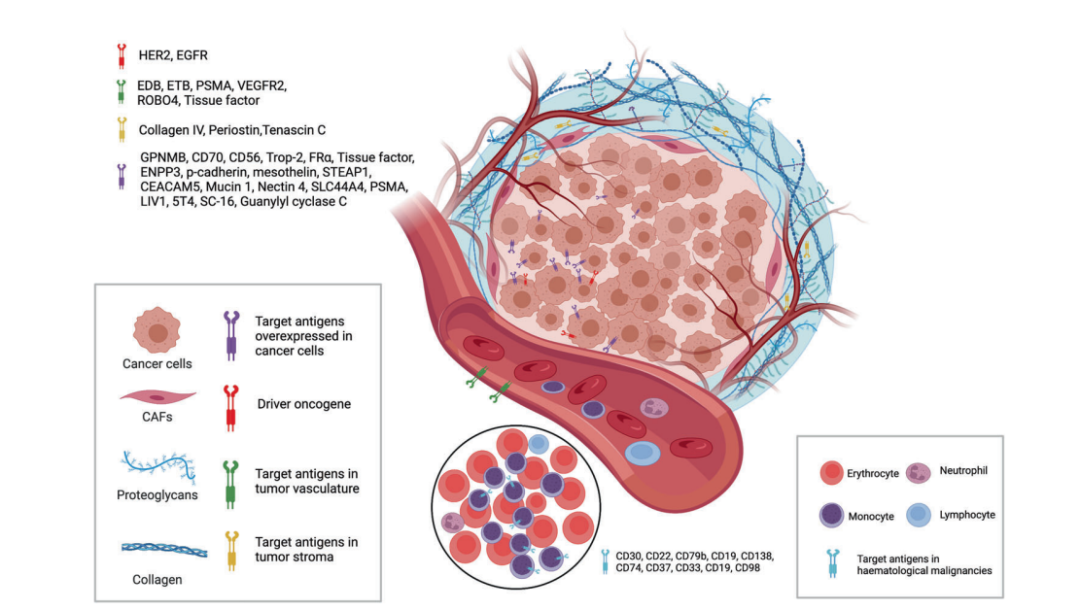
Figure 3. Targets Available From Tumor Cells and Tumor Microenvironment for ADCs
Currently, as shown in the figure above, the target antigens of approved ADC drugs are typically specific proteins that are overexpressed in cancer cells, including HER2, trop2, nectin4, and EGFR in solid tumors, as well as CD19, CD22, CD33, CD30, BCMA, and CD79b in hematological malignancies. Driven by fundamental research in oncology and immunology, the selection of ADC target antigens is gradually expanding from traditional tumor cell antigens to targets within the tumor microenvironment, such as the stroma and vascular system. Emerging preclinical and clinical evidence suggests that components of the neovascular system, subendothelial extracellular matrix, and tumor stroma may serve as valuable target antigens for ADC drug development.
Selection of Antibodies for ADCs
Targeting antibodies are crucial for the specific binding between the target antigens and ADCs. In addition to having high binding affinity for target antigens, ideal antibody fragments should promote effective internalization, exhibit low immunogenicity, and have a long plasma half-life.
In the early stages of ADC drug development, mouse-derived antibodies were primarily used, but due to severe immunogenicity-related side effects, the failure rate was high. With the advent of gene recombination technology, mouse-derived antibodies have mostly been replaced by chimeric antibodies and humanized antibodies. Currently, ADCs increasingly utilize fully humanized antibodies with significantly reduced immunogenicity. Most antibodies used in ADC drugs are immunoglobulin G (IgG) antibodies, including four subclasses: IgG1, IgG2, IgG3, and IgG4.
-
IgG1 is the commonly used antibody subclass for ADCs because it is the most abundant in serum and can induce strong effector functions through high binding affinity to Fc receptors, such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). These Fc-mediated effector functions play a crucial role in the anticancer activity of antibody drugs.
-
Due to rapid clearance, IgG3 is rarely used in ADCs. Unlike the other three subclasses with a half-life of approximately 21 days, IgG3 has a half-life of only about 7 days in serum.
-
IgG2 often exhibits a tendency to form dimers and aggregates in vivo, leading to decreased concentrations of ADC drugs.
-
IgG4 can induce ADCP; however, IgG4 is an abnormally dynamic antibody with Fab arm exchange, leading to reduced potency and ineffective targeting.
Regarding the internalization of the antibody-antigen complex, efficiency primarily depends on the binding affinity between the antibody and the tumor cell surface antigen, with higher affinity typically leading to faster internalization. However, antibodies with high antigen affinity may conversely reduce penetration into solid tumors. Treatment of solid tumors is more complex than that of hematological tumors because of the presence of a “binding site barrier (BSB)” in solid tumors, where the extremely strong binding between the antibody and antigen leads to ADCs being trapped near blood vessels after extravasation, with limited penetration into tumor cells far from the vessels. Therefore, the reasonable affinity between the antigen and antibody should be optimized to balance rapid uptake in target cells and anticancer efficacy.
In addition to binding affinity, another factor affecting tumor penetration is the size of the antibody. The large molecular weight of IgG antibodies (approximately 150 kDa) often limits their ability to permeate through capillaries and stroma in tumor tissues. Therefore, early ADCs primarily targeted hematological malignancies. To better apply ADCs in the treatment of solid tumors, researchers have attempted to miniaturize antibodies by removing the Fc fragment. Miniaturized antibodies not only retain high affinity and specificity but also penetrate solid tumors more easily through blood vessels, significantly enhancing their cytotoxic effects on solid tumors. However, this change can lead to a shortened half-life in vivo. Therefore, a comprehensive consideration of various factors should be made to achieve reasonable optimization when designing ADCs with miniaturized antibodies.
Selection of Linkers for ADCs
The linker in ADCs connects the antibody to the cytotoxic drug. It is one of the key factors related to ADC stability and the release curve of the payload, thus playing an important role in the final therapeutic indicators of ADCs.
Ideally, the linker should not induce ADC aggregation, should limit the premature release of the payload in plasma, and facilitate the release of the active drug at the desired targeting site. Depending on the metabolic pathways in cells, most ADC drugs utilize two types of linkers, including cleavable linkers and non-cleavable linkers. Cleavable linkers utilize environmental differences between systemic circulation and tumor cells to accurately release free cytotoxic drugs and can be further divided into chemical cleavable linkers (hydrazone bonds and disulfide bonds) and enzymatically cleavable linkers (glucuronide bonds and peptide bonds). Hydrazone is a typical acid-sensitive (pH-sensitive) linker.
Cleavable linkers take advantage of environmental differences between systemic circulation and tumor cells to accurately release free cytotoxic drugs and can be further divided into chemical cleavable linkers (hydrazone bonds and disulfide bonds) and enzymatically cleavable linkers (glucuronide bonds and peptide bonds). Another type of chemically sensitive cleavable linker is sensitive to reducing glutathione (GSH). GSH plays a crucial role in maintaining intracellular redox balance during cellular survival, proliferation, and differentiation. The concentration of GSH in blood is much lower than its intracellular concentration in cancer cells. Therefore, this type of linker can remain stable in the bloodstream while specifically releasing active payloads within cancer cells.
Peptide-based linkers are sensitive to lysosomal proteases and have been used in many ADCs. Lysosomal proteases, such as cathepsin B, are often overexpressed in cancer cells, allowing for the accurate release of drugs near tumors. Additionally, due to the presence of protease inhibitors in the bloodstream, enzymatically cleavable linkers are typically stable in systemic circulation, thus reducing various risks associated with premature cleavage. Among the approved ADC drugs, 9 out of 14 use peptide-based linkers.
Non-cleavable linkers (e.g., thioether or maleimide-based) are inert to the common chemical and enzymatic environments in vivo. The main advantage of non-cleavable linkers, due to improved plasma stability, is their lower off-target toxicity, but the bystander effect of the payload is affected.
Selection of Payloads for ADCs
Cytotoxic payloads are the warheads that exert cytotoxic effects after ADCs are internalized into cancer cells. Since only about 2% of ADCs can reach the targeted tumor site after intravenous administration, high-potency payloads (with IC50 in the nM and pM range) are critical for ADC development. Furthermore, these compounds should remain stable under physiological conditions and possess available functional groups for binding to antibodies.
Currently, the cytotoxic payloads used in ADCs primarily include potent microtubule inhibitors, DNA damaging agents, and immunomodulators (Table 1).
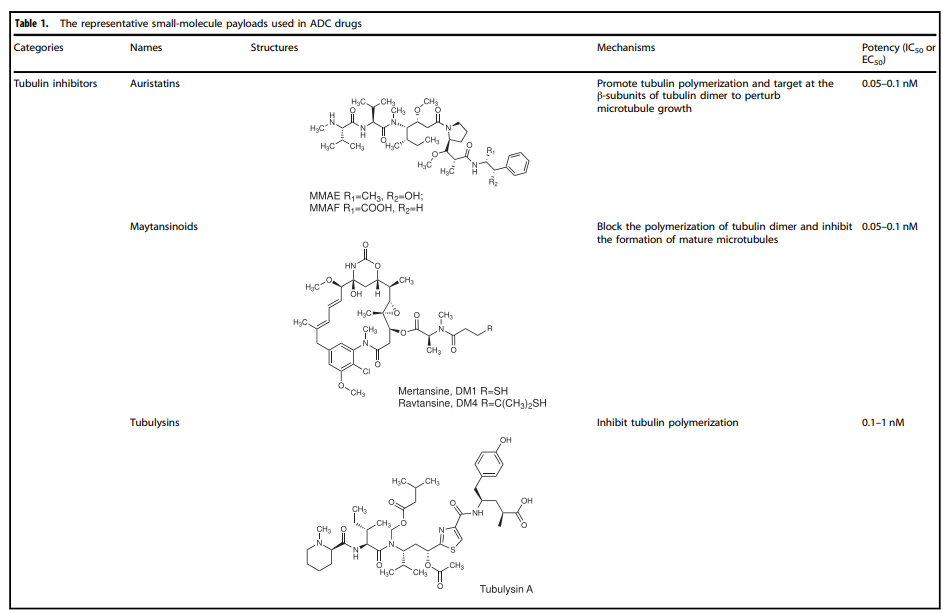
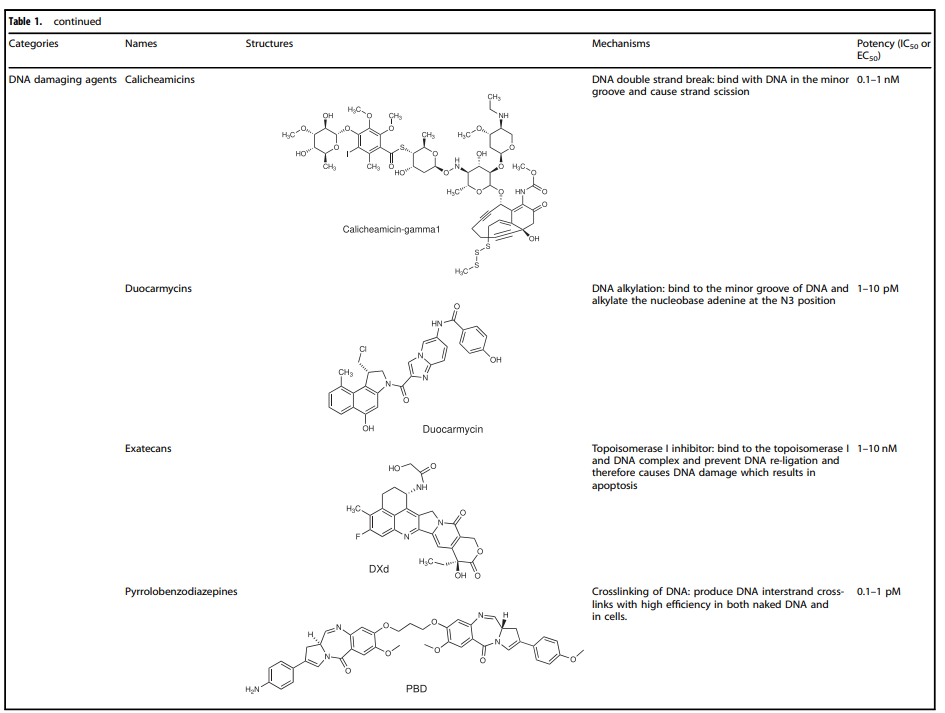
Table 1. Representative Small Molecule Payloads Used in ADC Drugs
Microtubule Disruptors
Microtubules are a major component of the cytoskeleton and play a critical role in cell division, particularly during the rapid proliferation of tumor cells. Microtubule inhibitors have become a class of anticancer drugs. Auristatins are important payloads used in ADCs, with monomethyl auristatin E (MMAE) or monomethyl auristatin F (MMAF) becoming mainstream payloads for ADC development. Among the 14 approved ADC drugs, 5 use MMAE/MMAF as payloads.
Maytansinoid Derivatives (DM1 and DM4)
Maytansine is a very effective microtubule assembly inhibitor that can induce cell cycle arrest. However, this structure is difficult to conjugate due to the lack of reactive functional groups. To overcome this issue, a series of highly effective derivatives containing SMe groups have been created. The first examples of this class of molecules are DM1 and DM4, which contain a methylthioacetyl group instead of the natural N-acetyl group.
The FDA approved ado-trastuzumab emtansine (TDM-1, KADCYLA®) in 2013 as the first ADC drug conjugated with maytansinoid derivatives. Additionally, microtubule inhibitors (microtubule inhibitors A-D, isolated from slime molds) represent another class of microtubule assembly inhibitors that exhibit good anticancer activity. For example, EC1169, a microtubule toxin B hydrazone conjugate targeting prostate-specific membrane antigen (PSMA), is currently being tested clinically (NCT02202447).
DNA Damaging Agents
Compared to the IC50 values of microtubule inhibitors in the nmol range (half-maximal inhibitory concentration), the IC50 values of DNA damaging agents can reach the pmol level, making ADCs conjugated with DNA damaging agents sometimes more effective and potentially able to work independently of the cell cycle (unlike microtubule inhibitors that primarily work during mitosis), and they can even be used for cells with low antigen expression.
The detailed mechanisms involved with DNA damaging agents mainly include: (i) DNA double-strand breaks, such as calicheamicin; (ii) DNA alkylation, such as duocarmycin; (iii) DNA intercalation, such as topoisomerase I inhibitors; (iv) DNA crosslinking, such as pyrrolobenzodiazepines (PBD).
It is precisely the various functions of the payload itself that inhibit tumor cell growth or directly kill tumor cells that enrich the mechanisms of action of ADC drugs.
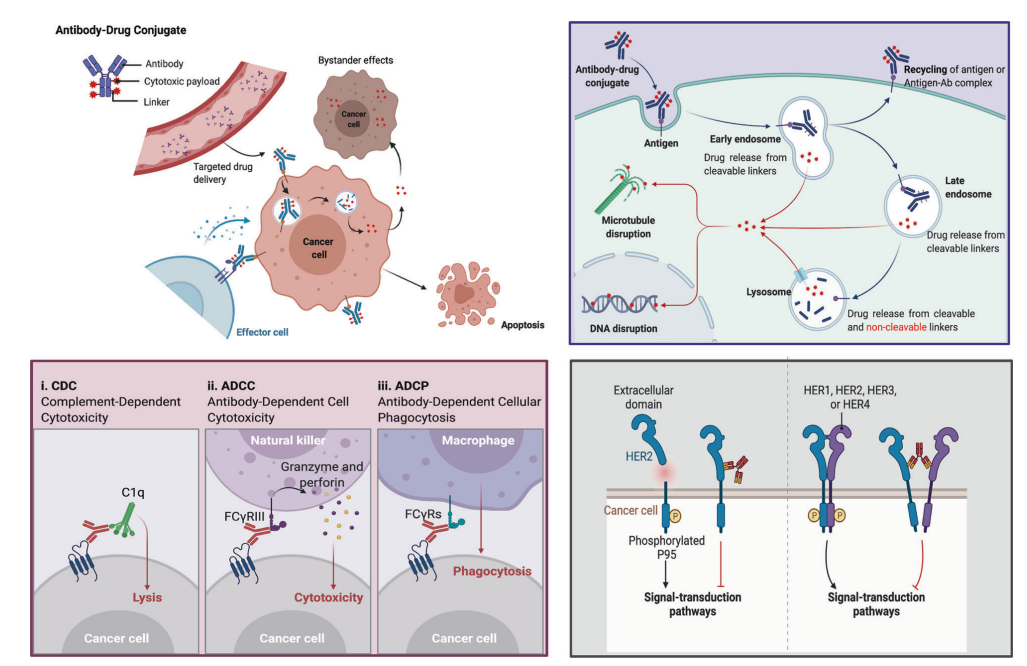
Figure 4. Overview of Mechanisms by Which ADCs Kill Cancer Cells
Upper right: The main core mechanisms of ADCs; lower left: The antibody component of ADCs binds to immune effector cells, triggering antitumor immunity, including CDC, ADCC, and ADCP effects; lower right: The antibody component of ADCs retains its active characteristics, thus interfering with target function and inhibiting downstream signaling that suppresses tumor growth.
Even with numerous antitumor mechanisms originating from the tumor-targeting elimination effect of ADCs, the cytotoxic effects received by the antibody, and the various antitumor mechanisms of the payload, the key to ADC success lies in the conjugation method of the antibody, payload, and linker.
Conjugation Methods
The site of linker conjugation and the conjugation method determine the drug-to-antibody ratio (DAR) and significantly affect the stability of the ADC as well as its pharmacokinetic and pharmacodynamic characteristics—high drug load often leads to rapid plasma clearance, while low DAR ADCs exhibit weaker activity.
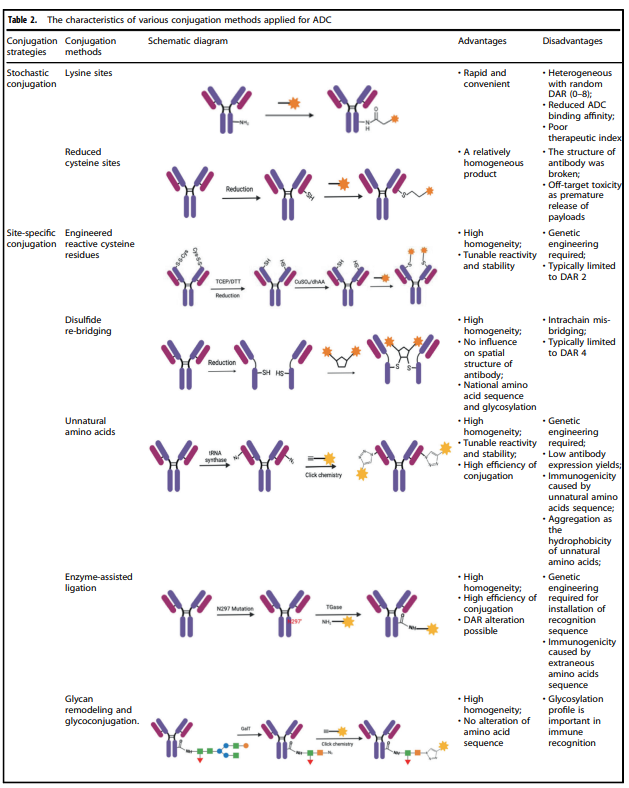
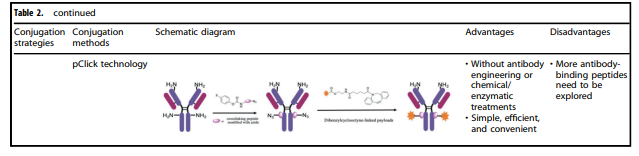
Table 2. Characteristics of Various Conjugation Methods Applied to ADCs
Table 2 summarizes the characteristics of various conjugation methods currently used for ADCs. Among them, chemical conjugation and enzymatic conjugation are the two most commonly used methods for linking antibody and payload components.
Chemical Conjugation
Chemical conjugation involves coupling the handle portion of the linker with the amino acid residues on the antibody’s surface, avoiding the complexity of determining suitable mutation sites and the potential challenges of scaling up and optimizing cell culture. Early chemical conjugation methods had great randomness in DAR and conjugation sites, resulting in significant variability in the produced ADCs and insufficient uniform quality.
Enzymatic Conjugation
By using genetically encoded amino acid tags inserted into the antibody sequence, the attachment of the payload can be achieved in a highly selective manner. These tags are specifically chosen to be recognized by enzymes capable of performing site-specific conjugation, such as formylglycine-generating enzyme (FGE), microbial transglutaminase (MTG), sortase enzymes, or tyrosinase.
Potential Development Directions for ADCs
With the continuous optimization of conjugation technologies and advancements in antibody engineering technologies, more ADC products targeting other antigens are under clinical exploration. For example, Trop2 ADC, Claudin18.2 ADC; among them, PD-L1 ADCs are also being explored by several companies.
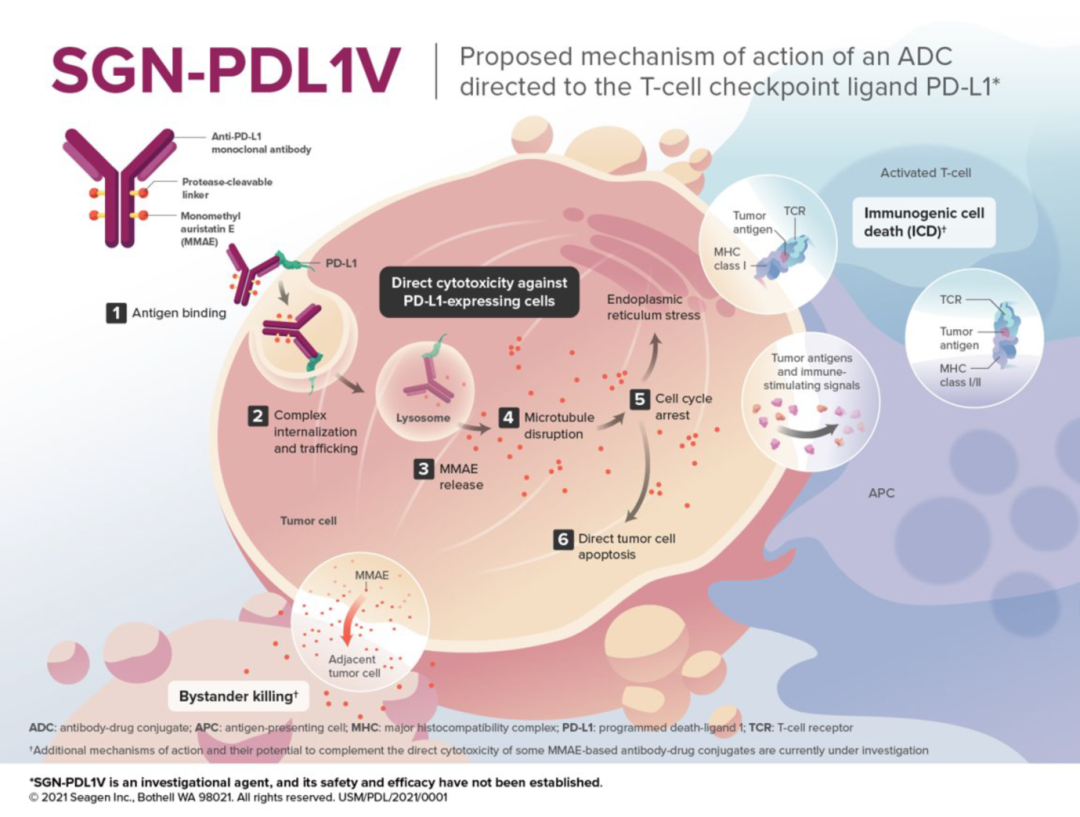
Figure 5. Mechanism of SGN-PDL1V (Source:2021 SITC)
For example, SGN-PDL1V, a novel ADC product targeting PD-L1 in the early stages of development in Seagen’s pipeline, utilizes MMAE as the payload. MMAE (monomethyl auristatin E) is the most widely used payload in ADCs, accounting for approximately 30% of the total, primarily exerting effective mitotic inhibition by inhibiting microtubule polymerization.
ADCs show drug concentrations in tumor tissues exceeding 100 times, greatly expanding the therapeutic window of cytotoxic agents, effectively reducing the side effects caused by systemic chemotherapy, thereby enabling the exploration of ADCs in conjunction with other cancer treatment modalities.
Additionally, the linker of SGN-PDL1V is protease-cleavable. The antibody is a fully human anti-PD-L1 monoclonal antibody (Seagen PD-L1 monoclonal antibody). The Seagen PD-L1 monoclonal antibody utilizes a human IgG1-Fc scaffold, engineered to eliminate effector functions of the Fc region, including complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP).
Moreover, considering that single-target antibodies often exhibit high rates of resistance, dual-target bispecific antibodies as ADCs are also being explored by various companies.
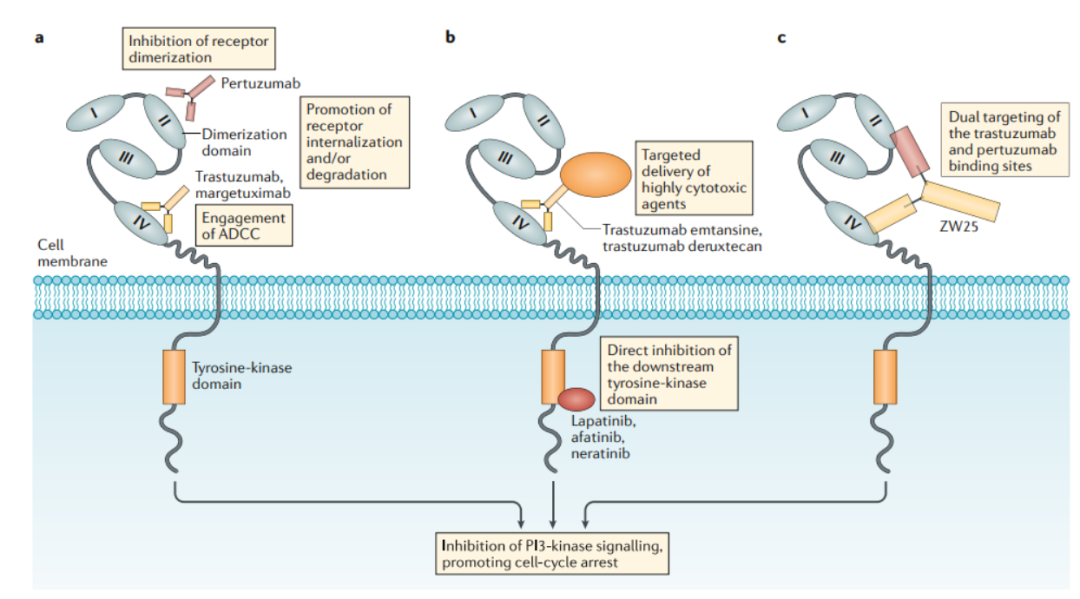
Figure 6. Mechanism of Action of HER2-Targeted Drugs
This field is still in the early stages of exploration, with the fastest progress being made by ZW-49, a bispecific antibody ADC developed by Zymeworks using its proprietary Azymetric™ Bispecifics and ZymeLink™ ADCs platform. It can specifically bind to two non-overlapping epitopes of the HER2 receptor, namely the binding sites of pertuzumab and trastuzumab. The goal is to achieve good therapeutic effects in clinical applications for patients resistant to trastuzumab, pertuzumab, or even new ADC drugs like TDM-1.
We look forward to more new ADCs being commercialized in the future, benefiting a broader range of patients in the fields of solid tumors and hematological malignancies.
References:
1. Signal Transduction and Targeted Therapy (2022) 7:93
2. Bioorg Chem. 2021 Nov;116:105366.
3. Oh DY, et al. Nat Rev Clin Oncol. 2019;10.1038/s41571-019-0268-3.
4. Mark D. Pegram, et al. Mol Cancer Ther; 20(8) August 2021.
