
This conference is themed “Innovative Processes, Achieving the Future of the Pharmaceutical Industry,” featuring one keynote session and three parallel sessions, gathering over 50 professional speakers from the fields of antibodies, ADCs, GLP-1, and peptides. It focuses on downstream processes and advanced separation and purification techniques, discussing the future of the pharmaceutical industry!

Known as “magic bullets,” ADCs have gained immense popularity in recent years. The design concept of this type of drug is clear: to utilize the high specificity of antigen-antibody interactions to selectively deliver toxins to antigen-positive cells, thereby selectively killing target cells and reducing damage to normal cells. However, like other drugs, the actual performance of ADCs in real battlefields may not always align with the design concept.
As more data emerges, the true working mechanism of successful ADCs becomes clearer. These are very complex drugs, so no single study can fully describe the mechanisms of ADCs. Like the story of the blind men and the elephant, we need to integrate various research findings to obtain a relatively complete picture.
In this article, I will list some important recent data points, allowing readers to connect these points and see what kind of anti-cancer drugs they can visualize.
1 ADCs Are Evolving from Molecular Targeted Delivery to Tissue TargetingADCs are essentially prodrugs, which are inactive by themselves (except for antibody-mediated efficacy) and only become lethal after the intracellular release of toxins. This distribution order is opposite to that of traditional small molecule drugs (which first contact normal cells and then distribute to tumors), giving ADCs an inherent advantage in tumor killing. More importantly, this activation strategy makes targeted killing possible. If the target antigen is expressed sufficiently specifically, then ADCs carrying super killers only need very low doses to precisely kill tumor cells without disturbing the daily lives of normal cells.However, highly specific tumor antigens are rare and are somewhat expressed in normal tissues. Tumors are relatively small compared to the human body, so most antibodies at low doses are primarily absorbed by normal tissue targets. This is also why diagnostic antibody imaging agents usually need to be combined with antibodies; some people also use monoclonal antibodies to solve binding site barrier issues.Additionally, non-specific binding of antibodies and endocytosis are rampant, as antibodies cannot wander indefinitely in the system. Therefore, the body has evolved a complete set of antigen-independent antibody metabolic clearance mechanisms. ADCs are not exempt from this; they will also be degraded and cleared at specified times and locations. Another very important factor is that although tumor antigen expression levels may be high, the antigens that can come into contact with the drug are usually limited due to the incomplete vascular structure and tight junctions between cells in tumor tissues. Using antibody imaging agents to diagnose tumors has high practical value, but so far, with the exception of a few peptide imaging agents, antibody-based imaging agents have not been marketed for use.One reason is that tumor antigens themselves have limited specificity and significant background noise. Another important reason is that tumor tissues are too chaotic, making it difficult for macromolecules to enter. For example, trastuzumab imaging agents cannot visualize one-third of HER2-positive lesions, while accumulating significantly in liver, kidney, and spleen tissues that do not express HER2. One can imagine what would happen if the payload carried by trastuzumab were not an imaging agent but a therapeutic agent.The concealment of tumor antigens, combined with interference from normal tissue in target and off-target antibody binding, leads to the current insufficient navigation ability of ADCs, causing over 99% of ADCs to be unable to locate tumor tissues and being forced to release toxins in normal tissues. The essential difference between ADCs and small molecule prodrugs is that ADCs cannot be excreted through the kidneys due to their large molecular weight and must be degraded to release toxins.An ADC with a half-life of one week means that within that week, regardless of how many antigens it finds, it must release 50% of its toxins, similar to a city’s garbage station with limited capacity that is difficult to locate, while citizens cannot avoid garbage, making it hard to maintain city cleanliness. Even in the microenvironment, tumor cells are not always the only cells releasing toxins; many non-specific release mechanisms may be involved in the toxin release of ADCs, which also diminishes the therapeutic value of target-mediated effects.Therefore, highly toxic ADCs do not succeed. It is only after the emergence of less toxic agents like DXd, which can freely enter and exit cells, that ADCs have truly entered the mainstream. For membrane-permeable toxins, intracellular release does not equal intracellular residence; rather, it will diffuse to other cells in the microenvironment. If released in tumor tissues, it will extend its killing power to tumor cells without target antigen expression, known as bystander killing, thus enhancing efficacy.However, this property is a double-edged sword; toxins released in normal cells will also diffuse to other normal cells, causing a wider range of damage. You cannot expect to socialize tumor killing while capitalizing on normal tissue damage. Clinically, ADCs have relatively rare target and off-target tissue toxicity; the same toxin, regardless of the ADC targeting different antigens, has similar toxicity profiles and maximum tolerated doses, and all are closely related to the toxin itself, indicating that regardless of where the toxin is released, the final toxicity is approximately the same and will be socialized throughout the entire compartment. DXd ADCs cause hair loss, and MMAF ADCs exhibit ocular toxicity. Of course, it is possible that ADCs with target toxicity have been eliminated during development, but based on my understanding of preclinical evaluation systems, this possibility is not very high.The efficacy situation is similar; although some ADCs yield greater benefits in populations with high antigen expression, most ADCs show a weak correlation between response and target expression in most applications. Most ADCs do not have reliable predictive efficacy biomarkers. Currently, 15 ADC products have been launched globally, and only three require testing for target antigen to select patients. Some high-density antigens like HER2 may also express certain levels in negative tumors, allowing for some opportunities to enter cells, but normal tissues like kidneys can express HER2 at IHC 3+ levels. This is not targeted delivery; rather, it is treating normal tissues as human shields (of course, different toxins have different killing powers against different normal tissues). If magic bullets work by first hitting normal tissues and then targeting tumors, it would be better to directly inject DXd into the veins.Several theoretical possibilities could explain this outcome. One is that the diffusion speed of small molecule chemotherapeutics is usually much faster than the speed of cell killing. Traditional chemotherapeutic drugs are generally infused into a patient’s veins, but the drugs do not stay in the bloodstream for long; instead, they distribute to systemic tissues in a short time, so intravenous administration of chemotherapy cannot be called targeted blood drugs. Similarly, the toxins released intracellularly by ADCs usually diffuse into the microenvironment before killing the tumor cells, which is one reason why intra-tumoral injection of chemotherapy does not achieve better tumor control.If the cells releasing the toxins must be killed before the toxins can continue to kill other cells, then antigen-positive cells should be cleared first, and the remaining tumor cells should primarily be antigen-negative. However, in the DAISY clinical trial of Enhertu, patients who responded showed only a slight decrease in HER2 expression after progression, with many patients showing no change in HER2 levels and a few even showing an increase, indicating that DXd performed indiscriminate killing in the microenvironment.Two is that the binding of ADCs to antigens is only one step in achieving targeted delivery; other steps must also cooperate to realize the value of this delivery step. For example, endocytosis must be fast enough, able to enter lysosomes, linkers must be cleaved, and toxins must escape from lysosomes; otherwise, cells have a series of operational mechanisms to expel ADCs, such as FcRn-mediated antibody recycling. Therefore, the binding of ADCs to antigens is a relatively fragile marriage, and many trivial matters can render this unique event that can achieve selective delivery ineffective.In summary, although ADCs are designed as molecular-targeted delivery technologies, under the guidance of clinical therapeutic windows, they have evolved into a tissue-level delivery technology, with both efficacy and toxicity determined at the tissue level, governed by the tissue’s free toxin AUC or Cmax. Of course, membrane-impermeable toxins or non-degradable ADCs are exceptions, but such ADCs are becoming increasingly rare. This macro image has profound implications for ADC target selection and molecular design.2ADCs Are Not Magic Bullets, But Magic TriggersADCs are referred to as magic bullets, meaning that chemotherapeutic drugs linked to antibodies can accurately kill target cells. However, this is based on the assumption that antibodies can specifically bind to antigens, allowing toxins to selectively kill antigen-positive cells. In reality, the binding of antibodies to antigens is just a small step; if endocytosis is slow, then ADC drugs may dissociate from the antigens. Even if endocytosis is efficient, if the linker hydrolysis is too slow, ADCs may be expelled to the extracellular space by FcRn.Compared to normal tissues, tumors are just a tiny area, so ADCs spend more time interacting with normal cells, and normal cells also have mechanisms for endocytosing ADCs and releasing toxins, such as Fc receptors. Although individual normal cells may have lower efficiency in comparison to tumor cells due to not expressing target antigens, normal cells dominate in number. Additionally, the vascular structures of normal tissues are more intact than those of tumor tissues, allowing for better drug penetration. Therefore, only less than 1% of ADCs are released in tumors.Even if ADCs can release toxins within tumor cells, the ability to achieve selective killing still requires that toxins remain in target cells for a sufficient amount of time. Small molecules have strong diffusion capabilities, and tumor cells also express some efflux pumps, so for toxins like DXd that have good membrane permeability, regardless of where they are released, they must gather together to act collectively to kill tumor cells in the microenvironment. The working mechanism of toxins like DM1, which have poor membrane permeability, may differ.If we compare ADCs to paratroopers, non-membrane-permeable T-DM1 completes its mission by killing within the landing cells, while T-DXd lands in the enemy’s backyard and then acts collectively, allowing it to kill both antigen-positive and negative tumor cells until expelled by the body. As mentioned earlier, this mechanism in the tumor microenvironment results in bystander killing effects, while in normal tissues, it leads to off-target toxicity.Thus, what ADCs can genuinely achieve is a certain degree of selective toxin release in target-positive antigen cells, with their working mechanism resembling a magic trigger, meaning that under certain circumstances, they can pull the trigger in tumor cells; whether they can become magic bullets depends on how long the bullets fly and where they land.Two years ago, scientists from Zymeworks published a significant paper indicating that ADCs do not improve absolute safety compared to small molecule toxins, as the maximum tolerated dose (MTD) has not increased. Logically, if ADCs only shoot at terrorists, then bystanders should not worry about being accidentally harmed. However, the actual situation is that the proportion of accidental harm at the same firepower has not decreased compared to small molecule toxins. Not only has the number of bystander injuries not decreased, but they are also all from the same group of unfortunate bystanders, such as rapidly dividing blood cells, which are unfortunate victims regardless of whether facing ADCs or small molecule toxins.This core viewpoint was raised as early as 2015 by FDA scientists, stating that regardless of the target, the strength and toxicity profile of ADCs are fundamentally determined by the toxin. The reason is that although bullets are primarily shot from tumor cells, the duration of flight and where they cause damage are ultimately determined by the AUC of the toxin in each compartment.Although absolute safety has not improved, this does not mean that ADCs do not offer additional benefits. When the blood drug concentration of toxins reaches MTD, the toxin concentrations of ADCs and small molecule toxins in the tumor microenvironment may vary to different extents based on antigen expression levels, thus potentially improving absolute efficacy. It is not easy to study how much ADCs accumulate in tumors compared to small molecule toxins, especially in actual patient tumors.Efficacy cannot serve as a substitute indicator for toxin accumulation; first, different tumors have different sensitivities to the same toxin, and antigen-high expressing tumors may be more sensitive or more tolerant to the toxin. Second, it is challenging to eliminate the influence of the antibody component, as for high-expressing antigen tumors, the antibody itself may mediate some immune killing, making the combined effects difficult to quantify. This ties into the third factor, which is sustained release, as ADCs have a sustained release toxin mechanism, which also increases the therapeutic window.One method is to attach various imaging agents to antibodies to see where the imaging agents remain, thereby inferring possible toxin release mechanisms. However, these techniques are difficult to precisely target cell types. Some studies have indeed pinpointed cell types, but the results are sometimes awkward. For example, a trastuzumab imaging agent primarily binds to macrophages in HER2-positive solid tumors and does not target HER2, raising various possibilities as to why highly specific trastuzumab behaves like a bad partner.Of course, one can directly measure the drug concentrations within tumor tissues, but there are several confounding factors. First, if measuring total toxin concentration, one must consider the drug-protein binding rates in different tissues; for instance, some small molecule toxins like MMAE may accumulate in tumor tissues relative to blood but bind non-specifically to proteins, meaning that free drug does not accumulate. Moreover, small molecules easily escape from tumors, so even if many are released in tumors, rapid escape may lead to decreased AUC, which interferes with fast-killing or slow-dissociating (slow Koff) toxins.This year, scientists from AstraZeneca published data on drug release of Enhertu in mouse models, indicating that the AUC of DXd released by Enhertu in HER2 high-expressing tumors (N87, where each cell expresses 3.5 million HER2) was 493, while in HER2 low-expressing MB468 (where each cell expresses 4800 HER2 receptors), it was 156. From a navigation capability perspective, HER2 should be considered the ceiling for ADC targets; the threefold toxin accumulation in strongly positive tumors compared to nearly negative tumors reflects the limits of this delivery mechanism under the current ADC design framework.Although a threefold accumulation has not met many people’s expectations, overcoming thermodynamic distribution laws is not an easy task. The therapeutic window of chemotherapy is usually very narrow, and a threefold increase is still a significant improvement. Coupled with the auxiliary effect of the antibody component, especially in high-expressing antigen populations, and the sustained release mechanism of ADC toxins, Enhertu significantly improves therapeutic outcomes compared to both trastuzumab and irinotecan.However, ADCs are also a class of drugs that are highly complex in design, production, and development. Whether this threefold accumulation justifies such a complicated design and whether it can be achieved through simpler designs is worth reconsidering. If one is willing to bear the high costs associated with antibody conjugation, we need to rethink how to genuinely achieve significant advantages over simpler molecular building models in flooding the microenvironment. ADCs have reached the moment described by Deming, where survival isn’t mandatory; they must either reinvent themselves or be replaced by other models. Both choices present new opportunities.3IO-Chemotherapy, or Even IO-IO CombinationsFor designers of magic bullets, a more hurtful statement than calling their weapon a mere combination of gunpowder and gun handle is that the targeting and navigation systems are not as important as they think. However, truly successful ADCs indeed leverage the synergistic or additive effects of both the antibody and toxin components. This combination effect has been relied upon in tumor therapy throughout history and has a complex background.The biggest obstacle in tumor drug development is the heterogeneity of tumors. If every tumor cell had the same structure and function and interacted with drugs in the same way, curing advanced tumors might have already become a reality. Tumors exhibit both genetic heterogeneity, meaning each tumor cell has a different genome and expresses key proteins in varying structures and quantities; as well as spatiotemporal heterogeneity, where protein expression varies among tumor cells at different developmental stages and in different tumor regions. The infiltrative areas of drugs also differ across lesions, and the existence of tumor stem cells makes treating advanced tumors almost an impossible task.Because of tumor heterogeneity, most therapies can only kill a portion of tumors. Targeted therapies struggle to kill antigen-negative tumors, and immunotherapies without the support of immune police in the microenvironment are left powerless. Small molecules have poor selectivity, and macromolecules cannot effectively infiltrate the microenvironment. Even chemotherapy struggles to kill tumor stem cells and cannot effectively prevent tumor recurrence.Therefore, combination therapy is the universal strategy in almost all tumor treatments. Even relatively clean targeted therapies can only effectively control advanced tumors in a few specific scenarios when combined with chemotherapy; currently, the vast majority of treatment regimens involve chemotherapy. Most targeted therapies need to be combined with chemotherapy to truly demonstrate therapeutic value; any new treatment plan still needs to bear some resemblance to chemotherapy. Chemotherapy drugs can massively kill a group of die-hard elements, but at the cost of mistakenly harming some innocent bystanders. They can also create enough chaos between tumor cells and the tumor microenvironment, driving the gangsters out of their bunkers, thus facilitating more precise and gentle modern therapies to target and kill, providing a basis for efficacy.While the discovery and development of chemotherapy drugs focus on their killing ability, in actual patients, they may not necessarily achieve this through direct killing. Most chemotherapeutic drugs that show sufficient therapeutic windows in clinical settings do not directly kill tumor cells at tolerated doses because the tumors in patients grow much slower than those in laboratory settings. One hypothesis is that before chemotherapy drugs reach their killing doses, they can cause significant chaos within tumor cells, thereby activating the immune system to indirectly kill tumors.This is akin to a killer entering a bar where gangsters are gathering to clean them out; although he has combat capabilities, when outnumbered, he can create chaos in the bar, attracting police to raid these previously unnoticed criminal elements. It has long been rumored that chemotherapy has components of immunotherapy, and now IO-ADC combinations are also an important direction in clinical development.Antibody drugs rely even more on the immune system; most antibody drug targets are expressed at higher levels in tumor cells but do not necessarily participate in crucial tumor developmental decisions. Of course, being expressed at higher levels than normal cells is also a suspicious sign, possibly indicating more covert participation. Even targets like HER2, which are highly associated with tumor growth, have limited effects on tumors when HER2 antibodies block signaling pathways; more often, they exert effects through immune-mediated mechanisms such as ADCC and ADCP. Therefore, ADCs in the tumor microenvironment exhibit both antibody-mediated immune killing and direct killing by chemotherapy or indirectly mediated immune killing, at least in certain scenarios simulating a combination of chemotherapy and antibody drugs.Of course, some ADCs have lower MTDs, and the antibody components do not reach therapeutic doses, but successful ADCs achieve effective antibody doses. Some ADCs have antibody components that do not exhibit significant single-agent activity, but local drug combinations in the tumor microenvironment may still produce therapeutic effects. Studies dating back to the last century have shown that a drug can enhance the therapeutic effect of another drug even at doses far below therapeutic levels; this is an important theoretical basis for designing small molecule dual-target drugs, so the contribution of the ADC antibody component should not be easily overlooked.As previously mentioned, most ADCs show a weak correlation between response and antigen expression levels in most scenarios; different ADCs with the same toxin exhibit very similar toxicity profiles and toxicity spectra, indicating that target-mediated biological effects primarily manifest at the tissue level rather than at the cellular level. Some ADCs show certain target-related responses and efficacy, such as Enhertu, where patients with high HER2 expression respond better. However, it is difficult to distinguish whether this is due to higher tumor toxin release throughput or greater sensitivity to trastuzumab-mediated immune killing.Animal models show that toxin accumulation in HER2 high-positive tumors is several times higher than in tumors with nearly no expression, but patient tumors may be more complex, and results may differ. HER2 imaging agents can distinguish between positive and negative lesions to some extent, but the rates of false positives and false negatives are still high, failing to meet commercial application standards. Additionally, as previously mentioned, the accumulation of antibodies in antigen-positive cells does not equal the ability to release toxins, and even if released, it does not guarantee sufficient retention time.Enhertu has been approved for the treatment of all HER2-positive solid tumors, but there is a significant difference in response rates between HER2-positive breast cancer (70%) and HER2-positive pancreatic cancer (5%). This seems more related to the underlying efficacy of trastuzumab in these two types of tumors rather than the navigation ability of Enhertu, as both are HER2-positive. Of course, the sensitivity of the two types of tumors to DXd may differ, and their toxin release capabilities may also vary.Tumor treatment has always relied on combination therapies centered around chemotherapy; ADCs are the only class of drugs that incorporate both the broad-spectrum killing of chemotherapy and the targeted killing of antibodies. Therefore, ADCs are the only single-agent drugs capable of launching a three-dimensional war against tumors, aligning with many years of experience in combination therapies against cancer. Unfortunately, many antibody targets for ADCs have not yet resulted in marketed monoclonal antibodies; one reason may be that these antibodies also require suitable chemotherapy combinations to be effective, while marketed chemotherapy drugs either do not match the mechanism or have incompatible pharmacokinetics, leading to low combination hit rates.In fact, there are very few antibody drug targets with significant single-agent activity against solid tumors. I counted and found only EGFR, HER2, PDL1, CLDN18.2, VEGFR2, PDGRFα, EpCAM, and GD2 have marketed drugs, and most are relatively small products. Therefore, any antibody with some single-agent activity should be developed as an ADC to test its druggability. The popularity of ADCs has resulted in clinical validation of which toxins are quality partners for antibody combinations; for instance, DXd seems to perform well with multiple antibodies, at least under sustained release conditions. These valuable data provide technical support for rapidly validating new target ADCs.4Curtin-Hammett RuleA basic assumption in ADC design is that the binding of antibodies to target antigens will initiate a killing process. If ADCs only bind to targets, then ADCs will only kill cells expressing those targets; if only tumors express the targets, then ADCs will only kill tumor cells. However, in actual execution, this logical chain often breaks down. The human body does not have antigens that are only expressed on tumor cells, and tumor surface antigens are not all identifiable by ADC drugs. Normal cells that do not express antigens can also internalize large amounts of ADCs and release toxins, and even within the tumor microenvironment, how much ADC toxin is released through antigen-positive tumor cells remains highly questionable.These little dirty secrets that cannot be disclosed will be discussed gradually in the future. Today, I will discuss that even if non-tumor cells do not interfere with ADC binding and toxin release, ADCs must complete tumor-specific killing under the right conditions.Chemical reaction kinetics provide some theoretical foundations for predicting the final results of multi-step processes, and one rule called the Curtin-Hammett rule is particularly relevant for understanding the final outcome of ADCs killing tumor cells in this multi-step process. This rule, in chemical terms, states that the product ratios of different pathways in a chemical reaction are determined by the activation energies of each pathway, regardless of the concentrations of intermediates along each reaction pathway, provided the conversion rates between intermediates are fast.To help non-chemistry professionals understand, let me translate this into a real-life scenario. Imagine two restaurants, A and B, on a street, each employing a person to attract customers at their doors. The person at restaurant A is more eloquent, so 90% of the foot traffic is drawn to his restaurant. But does this mean restaurant A will definitely have higher sales than restaurant B? Not necessarily. If restaurant A has a dirty and chaotic dining environment, poor service, and subpar dishes, most customers will ultimately dine at restaurant B, which is right next door. Of course, if restaurant A is in Beijing and restaurant B is in Shanghai, this rule becomes invalid, corresponding to a slower conversion rate of intermediates in a chemical reaction.The binding of ADCs to target antigens operates on the same principle. Typically, the speed at which antibodies bind to antigens is very fast, often determined by the diffusion speed of the molecules; however, whether they can be internalized into cells depends on the biological functions of each antigen and the construction mode of the antibodies. Once inside the cell, it does not mean that the toxins can be unloaded immediately; they must reach a special department rich in degradation enzymes called lysosomes to unload, and the quantity and activity of tissue proteases in lysosomes differ among cells, leading to different hydrolysis speeds of linkers in different tumor cells.The parking time in lysosomes is limited; if the hydrolysis speed is too slow, ADC molecules may be transported outside the cell by antibody recycling mechanisms such as FcRn, requiring them to queue again to return to lysosomes. Proteases in lysosomes do not only degrade linkers; if your toxin resembles a polypeptide, it may also be treated as waste and discarded. Previously, there were attempts to use Velcade as an ADC toxin, but it was ultimately treated as a polypeptide and broken down in lysosomes.Even if your ADC successfully releases all toxins at the target cells, this does not mean the assassination mission is complete, and the bounty can be claimed. Because tumor killing is a slow process, the timing of toxin release and tumor cell killing does not align. After the toxins are released, they lose all contact with the antibodies and diffuse randomly, acting independently. Some toxins, due to their slow membrane permeability, may remain in tumor cells for extended periods, providing enough time to kill that tumor cell, but the cost is that they can only kill that particular tumor cell and cannot enter other cells.Currently, most ADC toxins can produce what is known as bystander killing effects, meaning they can freely enter and exit cell membranes, with the timescale for this process measured in seconds. However, tumor cell killing in vitro also requires several days, and patient tumors grow much slower, so even the most sensitive tumors in the clinic require several weeks to show a response.Therefore, the timing of toxin departure from tumor cells and tumor killing operates on completely different timescales. Besides extremely potent ligands or irreversible ligands (with very high dissociation constants Koff, requiring a long time to dissociate from targets), most small molecule toxins will scatter before executing their tasks, even when released at their intended destination.Like all targeted delivery technology platforms, ADCs also hope to maintain drug concentrations above systemic exposure levels in the tumor microenvironment for extended periods, but this violates the second law of thermodynamics. To deviate from thermodynamic equilibrium, continuous energy input is required, which is the binding energy between antibodies and antigens. However, this is only a necessary but insufficient condition; as analyzed above, the efficiency of this limited binding energy is influenced by many factors, and if used improperly, it can easily lead to financial ruin. Similarly, binding between targets and ADCs is also necessary but not sufficient, akin to how increasing marriage rates does not equate to increasing birth rates, as the latter is influenced by many other factors.5Magic Bullets or Just BulletsWhile ADCs are much larger and more complex than chemotherapeutic drugs, they are fundamentally still chemotherapeutic agents. Although the antibody component serving as the driver may play a covert protective role in certain situations, the primary killing still depends on the toxin component; after all, magic bullets are still bullets. Despite the concept of ADCs being around for a long time and quickly resulting in heavyweight products like T-DM1, ADCs did not receive significant attention from mainstream pharmaceutical companies until the emergence of Enhertu.In 2020, Roche’s CEO stated, “We have shifted our technology priorities,” indicating that Roche had failed to master the complexity of ADCs. Roche’s position in the cancer drug field is comparable to Apple’s in the smartphone industry; they had already launched two heavyweight ADCs, T-DM1 and Polivy, and when the CEO speaks, the industry listens. In retrospect, it can be said that everything was in place, except for a quality toxin, which turned out to be DXd. Daiichi Sankyo had developed camptothecin derivatives for many years and launched irinotecan. While irinotecan has good efficacy, it still falls short compared to paclitaxel and platinum-based drugs. Later, Daiichi Sankyo discovered a more active analog, exatecan, but it was also more toxic, and despite conducting numerous clinical trials, they could not find a usable indication. At that time, Daiichi Sankyo aimed to develop exatecan as a macromolecular conjugate prodrug and pushed a macromolecular prodrug called DE310 into clinical trials.This prodrug released exatecan very slowly, but it still could not achieve a sufficient therapeutic window. Later, ADCs came into the spotlight, and Daiichi Sankyo transferred their experience with DE310 to ADC design, including the GGFG linker used in DE310. Even the earliest patents for Enhertu included exatecan as the toxin:
ADCs Are Evolving from Molecular Targeted Delivery to Tissue TargetingADCs are essentially prodrugs, which are inactive by themselves (except for antibody-mediated efficacy) and only become lethal after the intracellular release of toxins. This distribution order is opposite to that of traditional small molecule drugs (which first contact normal cells and then distribute to tumors), giving ADCs an inherent advantage in tumor killing. More importantly, this activation strategy makes targeted killing possible. If the target antigen is expressed sufficiently specifically, then ADCs carrying super killers only need very low doses to precisely kill tumor cells without disturbing the daily lives of normal cells.However, highly specific tumor antigens are rare and are somewhat expressed in normal tissues. Tumors are relatively small compared to the human body, so most antibodies at low doses are primarily absorbed by normal tissue targets. This is also why diagnostic antibody imaging agents usually need to be combined with antibodies; some people also use monoclonal antibodies to solve binding site barrier issues.Additionally, non-specific binding of antibodies and endocytosis are rampant, as antibodies cannot wander indefinitely in the system. Therefore, the body has evolved a complete set of antigen-independent antibody metabolic clearance mechanisms. ADCs are not exempt from this; they will also be degraded and cleared at specified times and locations. Another very important factor is that although tumor antigen expression levels may be high, the antigens that can come into contact with the drug are usually limited due to the incomplete vascular structure and tight junctions between cells in tumor tissues. Using antibody imaging agents to diagnose tumors has high practical value, but so far, with the exception of a few peptide imaging agents, antibody-based imaging agents have not been marketed for use.One reason is that tumor antigens themselves have limited specificity and significant background noise. Another important reason is that tumor tissues are too chaotic, making it difficult for macromolecules to enter. For example, trastuzumab imaging agents cannot visualize one-third of HER2-positive lesions, while accumulating significantly in liver, kidney, and spleen tissues that do not express HER2. One can imagine what would happen if the payload carried by trastuzumab were not an imaging agent but a therapeutic agent.The concealment of tumor antigens, combined with interference from normal tissue in target and off-target antibody binding, leads to the current insufficient navigation ability of ADCs, causing over 99% of ADCs to be unable to locate tumor tissues and being forced to release toxins in normal tissues. The essential difference between ADCs and small molecule prodrugs is that ADCs cannot be excreted through the kidneys due to their large molecular weight and must be degraded to release toxins.An ADC with a half-life of one week means that within that week, regardless of how many antigens it finds, it must release 50% of its toxins, similar to a city’s garbage station with limited capacity that is difficult to locate, while citizens cannot avoid garbage, making it hard to maintain city cleanliness. Even in the microenvironment, tumor cells are not always the only cells releasing toxins; many non-specific release mechanisms may be involved in the toxin release of ADCs, which also diminishes the therapeutic value of target-mediated effects.Therefore, highly toxic ADCs do not succeed. It is only after the emergence of less toxic agents like DXd, which can freely enter and exit cells, that ADCs have truly entered the mainstream. For membrane-permeable toxins, intracellular release does not equal intracellular residence; rather, it will diffuse to other cells in the microenvironment. If released in tumor tissues, it will extend its killing power to tumor cells without target antigen expression, known as bystander killing, thus enhancing efficacy.However, this property is a double-edged sword; toxins released in normal cells will also diffuse to other normal cells, causing a wider range of damage. You cannot expect to socialize tumor killing while capitalizing on normal tissue damage. Clinically, ADCs have relatively rare target and off-target tissue toxicity; the same toxin, regardless of the ADC targeting different antigens, has similar toxicity profiles and maximum tolerated doses, and all are closely related to the toxin itself, indicating that regardless of where the toxin is released, the final toxicity is approximately the same and will be socialized throughout the entire compartment. DXd ADCs cause hair loss, and MMAF ADCs exhibit ocular toxicity. Of course, it is possible that ADCs with target toxicity have been eliminated during development, but based on my understanding of preclinical evaluation systems, this possibility is not very high.The efficacy situation is similar; although some ADCs yield greater benefits in populations with high antigen expression, most ADCs show a weak correlation between response and target expression in most applications. Most ADCs do not have reliable predictive efficacy biomarkers. Currently, 15 ADC products have been launched globally, and only three require testing for target antigen to select patients. Some high-density antigens like HER2 may also express certain levels in negative tumors, allowing for some opportunities to enter cells, but normal tissues like kidneys can express HER2 at IHC 3+ levels. This is not targeted delivery; rather, it is treating normal tissues as human shields (of course, different toxins have different killing powers against different normal tissues). If magic bullets work by first hitting normal tissues and then targeting tumors, it would be better to directly inject DXd into the veins.Several theoretical possibilities could explain this outcome. One is that the diffusion speed of small molecule chemotherapeutics is usually much faster than the speed of cell killing. Traditional chemotherapeutic drugs are generally infused into a patient’s veins, but the drugs do not stay in the bloodstream for long; instead, they distribute to systemic tissues in a short time, so intravenous administration of chemotherapy cannot be called targeted blood drugs. Similarly, the toxins released intracellularly by ADCs usually diffuse into the microenvironment before killing the tumor cells, which is one reason why intra-tumoral injection of chemotherapy does not achieve better tumor control.If the cells releasing the toxins must be killed before the toxins can continue to kill other cells, then antigen-positive cells should be cleared first, and the remaining tumor cells should primarily be antigen-negative. However, in the DAISY clinical trial of Enhertu, patients who responded showed only a slight decrease in HER2 expression after progression, with many patients showing no change in HER2 levels and a few even showing an increase, indicating that DXd performed indiscriminate killing in the microenvironment.Two is that the binding of ADCs to antigens is only one step in achieving targeted delivery; other steps must also cooperate to realize the value of this delivery step. For example, endocytosis must be fast enough, able to enter lysosomes, linkers must be cleaved, and toxins must escape from lysosomes; otherwise, cells have a series of operational mechanisms to expel ADCs, such as FcRn-mediated antibody recycling. Therefore, the binding of ADCs to antigens is a relatively fragile marriage, and many trivial matters can render this unique event that can achieve selective delivery ineffective.In summary, although ADCs are designed as molecular-targeted delivery technologies, under the guidance of clinical therapeutic windows, they have evolved into a tissue-level delivery technology, with both efficacy and toxicity determined at the tissue level, governed by the tissue’s free toxin AUC or Cmax. Of course, membrane-impermeable toxins or non-degradable ADCs are exceptions, but such ADCs are becoming increasingly rare. This macro image has profound implications for ADC target selection and molecular design.2ADCs Are Not Magic Bullets, But Magic TriggersADCs are referred to as magic bullets, meaning that chemotherapeutic drugs linked to antibodies can accurately kill target cells. However, this is based on the assumption that antibodies can specifically bind to antigens, allowing toxins to selectively kill antigen-positive cells. In reality, the binding of antibodies to antigens is just a small step; if endocytosis is slow, then ADC drugs may dissociate from the antigens. Even if endocytosis is efficient, if the linker hydrolysis is too slow, ADCs may be expelled to the extracellular space by FcRn.Compared to normal tissues, tumors are just a tiny area, so ADCs spend more time interacting with normal cells, and normal cells also have mechanisms for endocytosing ADCs and releasing toxins, such as Fc receptors. Although individual normal cells may have lower efficiency in comparison to tumor cells due to not expressing target antigens, normal cells dominate in number. Additionally, the vascular structures of normal tissues are more intact than those of tumor tissues, allowing for better drug penetration. Therefore, only less than 1% of ADCs are released in tumors.Even if ADCs can release toxins within tumor cells, the ability to achieve selective killing still requires that toxins remain in target cells for a sufficient amount of time. Small molecules have strong diffusion capabilities, and tumor cells also express some efflux pumps, so for toxins like DXd that have good membrane permeability, regardless of where they are released, they must gather together to act collectively to kill tumor cells in the microenvironment. The working mechanism of toxins like DM1, which have poor membrane permeability, may differ.If we compare ADCs to paratroopers, non-membrane-permeable T-DM1 completes its mission by killing within the landing cells, while T-DXd lands in the enemy’s backyard and then acts collectively, allowing it to kill both antigen-positive and negative tumor cells until expelled by the body. As mentioned earlier, this mechanism in the tumor microenvironment results in bystander killing effects, while in normal tissues, it leads to off-target toxicity.Thus, what ADCs can genuinely achieve is a certain degree of selective toxin release in target-positive antigen cells, with their working mechanism resembling a magic trigger, meaning that under certain circumstances, they can pull the trigger in tumor cells; whether they can become magic bullets depends on how long the bullets fly and where they land.Two years ago, scientists from Zymeworks published a significant paper indicating that ADCs do not improve absolute safety compared to small molecule toxins, as the maximum tolerated dose (MTD) has not increased. Logically, if ADCs only shoot at terrorists, then bystanders should not worry about being accidentally harmed. However, the actual situation is that the proportion of accidental harm at the same firepower has not decreased compared to small molecule toxins. Not only has the number of bystander injuries not decreased, but they are also all from the same group of unfortunate bystanders, such as rapidly dividing blood cells, which are unfortunate victims regardless of whether facing ADCs or small molecule toxins.This core viewpoint was raised as early as 2015 by FDA scientists, stating that regardless of the target, the strength and toxicity profile of ADCs are fundamentally determined by the toxin. The reason is that although bullets are primarily shot from tumor cells, the duration of flight and where they cause damage are ultimately determined by the AUC of the toxin in each compartment.Although absolute safety has not improved, this does not mean that ADCs do not offer additional benefits. When the blood drug concentration of toxins reaches MTD, the toxin concentrations of ADCs and small molecule toxins in the tumor microenvironment may vary to different extents based on antigen expression levels, thus potentially improving absolute efficacy. It is not easy to study how much ADCs accumulate in tumors compared to small molecule toxins, especially in actual patient tumors.Efficacy cannot serve as a substitute indicator for toxin accumulation; first, different tumors have different sensitivities to the same toxin, and antigen-high expressing tumors may be more sensitive or more tolerant to the toxin. Second, it is challenging to eliminate the influence of the antibody component, as for high-expressing antigen tumors, the antibody itself may mediate some immune killing, making the combined effects difficult to quantify. This ties into the third factor, which is sustained release, as ADCs have a sustained release toxin mechanism, which also increases the therapeutic window.One method is to attach various imaging agents to antibodies to see where the imaging agents remain, thereby inferring possible toxin release mechanisms. However, these techniques are difficult to precisely target cell types. Some studies have indeed pinpointed cell types, but the results are sometimes awkward. For example, a trastuzumab imaging agent primarily binds to macrophages in HER2-positive solid tumors and does not target HER2, raising various possibilities as to why highly specific trastuzumab behaves like a bad partner.Of course, one can directly measure the drug concentrations within tumor tissues, but there are several confounding factors. First, if measuring total toxin concentration, one must consider the drug-protein binding rates in different tissues; for instance, some small molecule toxins like MMAE may accumulate in tumor tissues relative to blood but bind non-specifically to proteins, meaning that free drug does not accumulate. Moreover, small molecules easily escape from tumors, so even if many are released in tumors, rapid escape may lead to decreased AUC, which interferes with fast-killing or slow-dissociating (slow Koff) toxins.This year, scientists from AstraZeneca published data on drug release of Enhertu in mouse models, indicating that the AUC of DXd released by Enhertu in HER2 high-expressing tumors (N87, where each cell expresses 3.5 million HER2) was 493, while in HER2 low-expressing MB468 (where each cell expresses 4800 HER2 receptors), it was 156. From a navigation capability perspective, HER2 should be considered the ceiling for ADC targets; the threefold toxin accumulation in strongly positive tumors compared to nearly negative tumors reflects the limits of this delivery mechanism under the current ADC design framework.Although a threefold accumulation has not met many people’s expectations, overcoming thermodynamic distribution laws is not an easy task. The therapeutic window of chemotherapy is usually very narrow, and a threefold increase is still a significant improvement. Coupled with the auxiliary effect of the antibody component, especially in high-expressing antigen populations, and the sustained release mechanism of ADC toxins, Enhertu significantly improves therapeutic outcomes compared to both trastuzumab and irinotecan.However, ADCs are also a class of drugs that are highly complex in design, production, and development. Whether this threefold accumulation justifies such a complicated design and whether it can be achieved through simpler designs is worth reconsidering. If one is willing to bear the high costs associated with antibody conjugation, we need to rethink how to genuinely achieve significant advantages over simpler molecular building models in flooding the microenvironment. ADCs have reached the moment described by Deming, where survival isn’t mandatory; they must either reinvent themselves or be replaced by other models. Both choices present new opportunities.3IO-Chemotherapy, or Even IO-IO CombinationsFor designers of magic bullets, a more hurtful statement than calling their weapon a mere combination of gunpowder and gun handle is that the targeting and navigation systems are not as important as they think. However, truly successful ADCs indeed leverage the synergistic or additive effects of both the antibody and toxin components. This combination effect has been relied upon in tumor therapy throughout history and has a complex background.The biggest obstacle in tumor drug development is the heterogeneity of tumors. If every tumor cell had the same structure and function and interacted with drugs in the same way, curing advanced tumors might have already become a reality. Tumors exhibit both genetic heterogeneity, meaning each tumor cell has a different genome and expresses key proteins in varying structures and quantities; as well as spatiotemporal heterogeneity, where protein expression varies among tumor cells at different developmental stages and in different tumor regions. The infiltrative areas of drugs also differ across lesions, and the existence of tumor stem cells makes treating advanced tumors almost an impossible task.Because of tumor heterogeneity, most therapies can only kill a portion of tumors. Targeted therapies struggle to kill antigen-negative tumors, and immunotherapies without the support of immune police in the microenvironment are left powerless. Small molecules have poor selectivity, and macromolecules cannot effectively infiltrate the microenvironment. Even chemotherapy struggles to kill tumor stem cells and cannot effectively prevent tumor recurrence.Therefore, combination therapy is the universal strategy in almost all tumor treatments. Even relatively clean targeted therapies can only effectively control advanced tumors in a few specific scenarios when combined with chemotherapy; currently, the vast majority of treatment regimens involve chemotherapy. Most targeted therapies need to be combined with chemotherapy to truly demonstrate therapeutic value; any new treatment plan still needs to bear some resemblance to chemotherapy. Chemotherapy drugs can massively kill a group of die-hard elements, but at the cost of mistakenly harming some innocent bystanders. They can also create enough chaos between tumor cells and the tumor microenvironment, driving the gangsters out of their bunkers, thus facilitating more precise and gentle modern therapies to target and kill, providing a basis for efficacy.While the discovery and development of chemotherapy drugs focus on their killing ability, in actual patients, they may not necessarily achieve this through direct killing. Most chemotherapeutic drugs that show sufficient therapeutic windows in clinical settings do not directly kill tumor cells at tolerated doses because the tumors in patients grow much slower than those in laboratory settings. One hypothesis is that before chemotherapy drugs reach their killing doses, they can cause significant chaos within tumor cells, thereby activating the immune system to indirectly kill tumors.This is akin to a killer entering a bar where gangsters are gathering to clean them out; although he has combat capabilities, when outnumbered, he can create chaos in the bar, attracting police to raid these previously unnoticed criminal elements. It has long been rumored that chemotherapy has components of immunotherapy, and now IO-ADC combinations are also an important direction in clinical development.Antibody drugs rely even more on the immune system; most antibody drug targets are expressed at higher levels in tumor cells but do not necessarily participate in crucial tumor developmental decisions. Of course, being expressed at higher levels than normal cells is also a suspicious sign, possibly indicating more covert participation. Even targets like HER2, which are highly associated with tumor growth, have limited effects on tumors when HER2 antibodies block signaling pathways; more often, they exert effects through immune-mediated mechanisms such as ADCC and ADCP. Therefore, ADCs in the tumor microenvironment exhibit both antibody-mediated immune killing and direct killing by chemotherapy or indirectly mediated immune killing, at least in certain scenarios simulating a combination of chemotherapy and antibody drugs.Of course, some ADCs have lower MTDs, and the antibody components do not reach therapeutic doses, but successful ADCs achieve effective antibody doses. Some ADCs have antibody components that do not exhibit significant single-agent activity, but local drug combinations in the tumor microenvironment may still produce therapeutic effects. Studies dating back to the last century have shown that a drug can enhance the therapeutic effect of another drug even at doses far below therapeutic levels; this is an important theoretical basis for designing small molecule dual-target drugs, so the contribution of the ADC antibody component should not be easily overlooked.As previously mentioned, most ADCs show a weak correlation between response and antigen expression levels in most scenarios; different ADCs with the same toxin exhibit very similar toxicity profiles and toxicity spectra, indicating that target-mediated biological effects primarily manifest at the tissue level rather than at the cellular level. Some ADCs show certain target-related responses and efficacy, such as Enhertu, where patients with high HER2 expression respond better. However, it is difficult to distinguish whether this is due to higher tumor toxin release throughput or greater sensitivity to trastuzumab-mediated immune killing.Animal models show that toxin accumulation in HER2 high-positive tumors is several times higher than in tumors with nearly no expression, but patient tumors may be more complex, and results may differ. HER2 imaging agents can distinguish between positive and negative lesions to some extent, but the rates of false positives and false negatives are still high, failing to meet commercial application standards. Additionally, as previously mentioned, the accumulation of antibodies in antigen-positive cells does not equal the ability to release toxins, and even if released, it does not guarantee sufficient retention time.Enhertu has been approved for the treatment of all HER2-positive solid tumors, but there is a significant difference in response rates between HER2-positive breast cancer (70%) and HER2-positive pancreatic cancer (5%). This seems more related to the underlying efficacy of trastuzumab in these two types of tumors rather than the navigation ability of Enhertu, as both are HER2-positive. Of course, the sensitivity of the two types of tumors to DXd may differ, and their toxin release capabilities may also vary.Tumor treatment has always relied on combination therapies centered around chemotherapy; ADCs are the only class of drugs that incorporate both the broad-spectrum killing of chemotherapy and the targeted killing of antibodies. Therefore, ADCs are the only single-agent drugs capable of launching a three-dimensional war against tumors, aligning with many years of experience in combination therapies against cancer. Unfortunately, many antibody targets for ADCs have not yet resulted in marketed monoclonal antibodies; one reason may be that these antibodies also require suitable chemotherapy combinations to be effective, while marketed chemotherapy drugs either do not match the mechanism or have incompatible pharmacokinetics, leading to low combination hit rates.In fact, there are very few antibody drug targets with significant single-agent activity against solid tumors. I counted and found only EGFR, HER2, PDL1, CLDN18.2, VEGFR2, PDGRFα, EpCAM, and GD2 have marketed drugs, and most are relatively small products. Therefore, any antibody with some single-agent activity should be developed as an ADC to test its druggability. The popularity of ADCs has resulted in clinical validation of which toxins are quality partners for antibody combinations; for instance, DXd seems to perform well with multiple antibodies, at least under sustained release conditions. These valuable data provide technical support for rapidly validating new target ADCs.4Curtin-Hammett RuleA basic assumption in ADC design is that the binding of antibodies to target antigens will initiate a killing process. If ADCs only bind to targets, then ADCs will only kill cells expressing those targets; if only tumors express the targets, then ADCs will only kill tumor cells. However, in actual execution, this logical chain often breaks down. The human body does not have antigens that are only expressed on tumor cells, and tumor surface antigens are not all identifiable by ADC drugs. Normal cells that do not express antigens can also internalize large amounts of ADCs and release toxins, and even within the tumor microenvironment, how much ADC toxin is released through antigen-positive tumor cells remains highly questionable.These little dirty secrets that cannot be disclosed will be discussed gradually in the future. Today, I will discuss that even if non-tumor cells do not interfere with ADC binding and toxin release, ADCs must complete tumor-specific killing under the right conditions.Chemical reaction kinetics provide some theoretical foundations for predicting the final results of multi-step processes, and one rule called the Curtin-Hammett rule is particularly relevant for understanding the final outcome of ADCs killing tumor cells in this multi-step process. This rule, in chemical terms, states that the product ratios of different pathways in a chemical reaction are determined by the activation energies of each pathway, regardless of the concentrations of intermediates along each reaction pathway, provided the conversion rates between intermediates are fast.To help non-chemistry professionals understand, let me translate this into a real-life scenario. Imagine two restaurants, A and B, on a street, each employing a person to attract customers at their doors. The person at restaurant A is more eloquent, so 90% of the foot traffic is drawn to his restaurant. But does this mean restaurant A will definitely have higher sales than restaurant B? Not necessarily. If restaurant A has a dirty and chaotic dining environment, poor service, and subpar dishes, most customers will ultimately dine at restaurant B, which is right next door. Of course, if restaurant A is in Beijing and restaurant B is in Shanghai, this rule becomes invalid, corresponding to a slower conversion rate of intermediates in a chemical reaction.The binding of ADCs to target antigens operates on the same principle. Typically, the speed at which antibodies bind to antigens is very fast, often determined by the diffusion speed of the molecules; however, whether they can be internalized into cells depends on the biological functions of each antigen and the construction mode of the antibodies. Once inside the cell, it does not mean that the toxins can be unloaded immediately; they must reach a special department rich in degradation enzymes called lysosomes to unload, and the quantity and activity of tissue proteases in lysosomes differ among cells, leading to different hydrolysis speeds of linkers in different tumor cells.The parking time in lysosomes is limited; if the hydrolysis speed is too slow, ADC molecules may be transported outside the cell by antibody recycling mechanisms such as FcRn, requiring them to queue again to return to lysosomes. Proteases in lysosomes do not only degrade linkers; if your toxin resembles a polypeptide, it may also be treated as waste and discarded. Previously, there were attempts to use Velcade as an ADC toxin, but it was ultimately treated as a polypeptide and broken down in lysosomes.Even if your ADC successfully releases all toxins at the target cells, this does not mean the assassination mission is complete, and the bounty can be claimed. Because tumor killing is a slow process, the timing of toxin release and tumor cell killing does not align. After the toxins are released, they lose all contact with the antibodies and diffuse randomly, acting independently. Some toxins, due to their slow membrane permeability, may remain in tumor cells for extended periods, providing enough time to kill that tumor cell, but the cost is that they can only kill that particular tumor cell and cannot enter other cells.Currently, most ADC toxins can produce what is known as bystander killing effects, meaning they can freely enter and exit cell membranes, with the timescale for this process measured in seconds. However, tumor cell killing in vitro also requires several days, and patient tumors grow much slower, so even the most sensitive tumors in the clinic require several weeks to show a response.Therefore, the timing of toxin departure from tumor cells and tumor killing operates on completely different timescales. Besides extremely potent ligands or irreversible ligands (with very high dissociation constants Koff, requiring a long time to dissociate from targets), most small molecule toxins will scatter before executing their tasks, even when released at their intended destination.Like all targeted delivery technology platforms, ADCs also hope to maintain drug concentrations above systemic exposure levels in the tumor microenvironment for extended periods, but this violates the second law of thermodynamics. To deviate from thermodynamic equilibrium, continuous energy input is required, which is the binding energy between antibodies and antigens. However, this is only a necessary but insufficient condition; as analyzed above, the efficiency of this limited binding energy is influenced by many factors, and if used improperly, it can easily lead to financial ruin. Similarly, binding between targets and ADCs is also necessary but not sufficient, akin to how increasing marriage rates does not equate to increasing birth rates, as the latter is influenced by many other factors.5Magic Bullets or Just BulletsWhile ADCs are much larger and more complex than chemotherapeutic drugs, they are fundamentally still chemotherapeutic agents. Although the antibody component serving as the driver may play a covert protective role in certain situations, the primary killing still depends on the toxin component; after all, magic bullets are still bullets. Despite the concept of ADCs being around for a long time and quickly resulting in heavyweight products like T-DM1, ADCs did not receive significant attention from mainstream pharmaceutical companies until the emergence of Enhertu.In 2020, Roche’s CEO stated, “We have shifted our technology priorities,” indicating that Roche had failed to master the complexity of ADCs. Roche’s position in the cancer drug field is comparable to Apple’s in the smartphone industry; they had already launched two heavyweight ADCs, T-DM1 and Polivy, and when the CEO speaks, the industry listens. In retrospect, it can be said that everything was in place, except for a quality toxin, which turned out to be DXd. Daiichi Sankyo had developed camptothecin derivatives for many years and launched irinotecan. While irinotecan has good efficacy, it still falls short compared to paclitaxel and platinum-based drugs. Later, Daiichi Sankyo discovered a more active analog, exatecan, but it was also more toxic, and despite conducting numerous clinical trials, they could not find a usable indication. At that time, Daiichi Sankyo aimed to develop exatecan as a macromolecular conjugate prodrug and pushed a macromolecular prodrug called DE310 into clinical trials.This prodrug released exatecan very slowly, but it still could not achieve a sufficient therapeutic window. Later, ADCs came into the spotlight, and Daiichi Sankyo transferred their experience with DE310 to ADC design, including the GGFG linker used in DE310. Even the earliest patents for Enhertu included exatecan as the toxin:
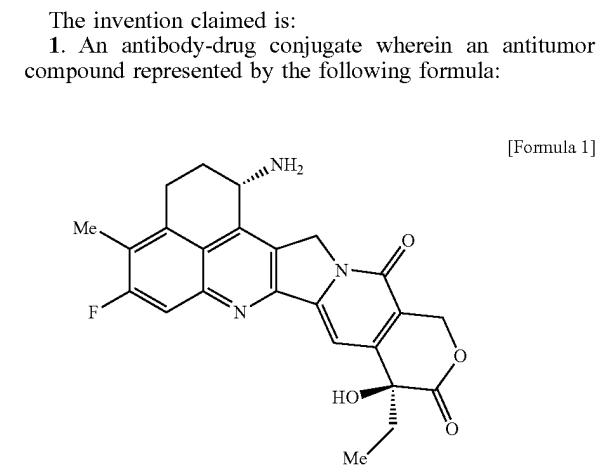
Although exatecan is more active than SN38, it is still considered a relatively weak toxin for ADCs, a shortcoming that can be addressed with a high DAR value. Exatecan has an amino group that can directly couple with GGFG via an amide bond, but this coupling can result in some cleavage at the G-F bond during enzyme hydrolysis, releasing G-exatecan. Considering some physicochemical properties, Daiichi Sankyo scientists added a hydroxyethylamine group to exatecan, resulting in the now-famous DXd (deruxtecan).Before this, how GGFG could couple with hydroxyl compounds was already known technology, so making Enhertu was not complicated. DXd is approximately five times weaker than exatecan, which does not align with the mainstream ADC design philosophy at the time. However, no one cares about DXd’s in vitro activity issues anymore.The success of Enhertu has some unexpected elements, as can be seen from Daiichi Sankyo’s stock over the past 20 years. As for why Enhertu is so successful, especially in HER2 ultralow breast cancer, the industry is still researching, but it is estimated that DXd possesses some unique superpowers. Currently, at least 10 other HER2 ADC products using different toxins have failed in clinical trials, and other more lethal agents like CAR-T, CAR-M, and immunotoxins have also met with failure. Meanwhile, the five different target ADCs using DXd are all highly valuable, with total transaction amounts of $35 billion being transferred to AstraZeneca and Merck.As for the mysterious abilities of DXd, it may take a long time to clarify, but it could simply be that it has a higher safety window than exatecan and is itself a quality small molecule chemotherapeutic agent. Unfortunately, DXd has never entered clinical research, so there are no head-to-head comparison data. It may also produce some immune activation effects at non-killing doses or directly induce ICD (immunogenic cell death) or enhance the ADCC of antibodies and other immune effects.DXd has a relatively short half-life, which is a disadvantage for small molecule drugs but an advantage for ADC toxins, as the toxins entering the bloodstream are quickly eliminated by metabolic organs like the liver and kidneys, maintaining a drug gradient between tumor tissues and the blood system. Perhaps it is this combination of factors that makes it difficult to judge with the currently limited public data.New drug discovery is a super complex system aimed at reducing clinical failure risks through a series of so-called de-risking screening processes, avoiding the trial-and-error approach in clinical trials. Unfortunately, truly pioneering new drugs are still primarily filtered through clinical trials, and rational drug design remains a gamble. Even so-called me-too follow-up drugs may be easier, but it depends on the specific situation; for instance, important drugs like paclitaxel, proton pump inhibitors, thalidomide-like molecular gels, and P2Y12 receptor agonists have me-too drugs, but they only have very slight structural differences from the pioneering drugs.This indicates that the mechanism alone is insufficient to filter out quality drugs; otherwise, someone would have already screened for new structural types. Many mainstream anticancer drugs that have been used for decades still do not have a clear understanding of their true working mechanisms; for example, paclitaxel does not achieve sufficient intracellular concentrations to inhibit microtubule protein dissociation at clinically effective doses, and other important drugs like platinum-based agents and tamoxifen are similar. DXd is just the latest case in this ongoing mystery; the more such mysteries there are, the better.6Who Is Casting the Spell?ADCs are inactive by themselves (except for antibody-mediated immune killing); only the release of toxins can confer tumor-killing activity. However, the details of toxin release in vivo, especially within patients, remain somewhat murky. While the design of ADCs aims for toxin release through internalization by antigen-positive tumor cells, proteolytic enzymes exist not only in tumor cells but also are not primarily sourced from them. The linkers of ADCs primarily undergo hydrolysis through tissue proteases but may also become substrates for other proteases. Thus, who is behind the magic is not a clear-cut issue.Although antibody drugs have a history of 30 years, our understanding of the degradation processes of antibodies in the human body still has many gaps. Isotope labeling studies have shown that the organs with the highest metabolic degradation efficiency for antibodies in mice are the liver, spleen, and kidneys; however, due to the larger amounts of skin and muscle tissue, the absolute amount of antibody protein degraded primarily occurs in the liver, skin, and muscle, clearing approximately 75% of antibodies. Many patients with advanced tumors experience significant weight loss, and there is a hypothesis that the mechanisms of muscle degradation may be linked to increased antibody degradation.It is generally believed that the endothelial phagocytic system is the primary mechanism for antibody degradation, with monocytes and macrophages being the main cell types. These cells naturally perform the function of phagocytosing and degrading waste proteins and other biological materials, so this system has a high throughput and strong internalization and degradation capabilities, which are also required for the release of ADC toxins.In the tumor microenvironment, there are also numerous tumor-associated macrophages (TAMs), which are usually co-opted by tumors to become accomplices in tumor growth. TAMs are highly abundant and constitute the largest immune cell population in the microenvironment, sometimes outnumbering tumor cells in certain lesions. Unlike tumor cells, which are tightly linked in a structured manner, TAMs can move freely in the microenvironment, as these cells need to patrol their respective tissues to clear waste proteins before becoming accomplices.Macrophages seek out culprits based on chemical signals, which is why some researchers have used macrophages as carriers for chemotherapeutic drugs, utilizing their ability to recognize tumors or other abnormal tissues to deliver drugs to diseased tissues. Drugs can be loaded into macrophages in vitro or can adhere to macrophages in vivo due to their natural tendency to