Click the link at the end to read the original article directly.

Research Background


Single-atom catalysts (SACs) have garnered significant attention in the field of heterogeneous electrocatalysis due to their maximized atomic utilization and high catalytic activity. However, the actual catalytic environment is much more complex than ideal models. The catalyst surface undergoes dynamic reshaping under operational conditions due to applied potentials, electrolyte environments, and reaction intermediates/products, leading to continuous changes in the coordination geometry and electronic properties of active sites, and may even trigger the rearrangement, migration, or detachment of metal atoms, forming metal clusters. Therefore, understanding the active sites of SACs under real operating conditions is crucial. Ni−N−C catalysts exhibit high activity and selectivity in the CO₂ reduction reaction (CO₂RR), but the nature of their true active sites remains unclear. Furthermore, the origin of their high CO₂RR selectivity cannot be satisfactorily explained by traditional static models, and the phenomenon of decreased CO selectivity at more negative potentials also lacks a clear mechanistic explanation.
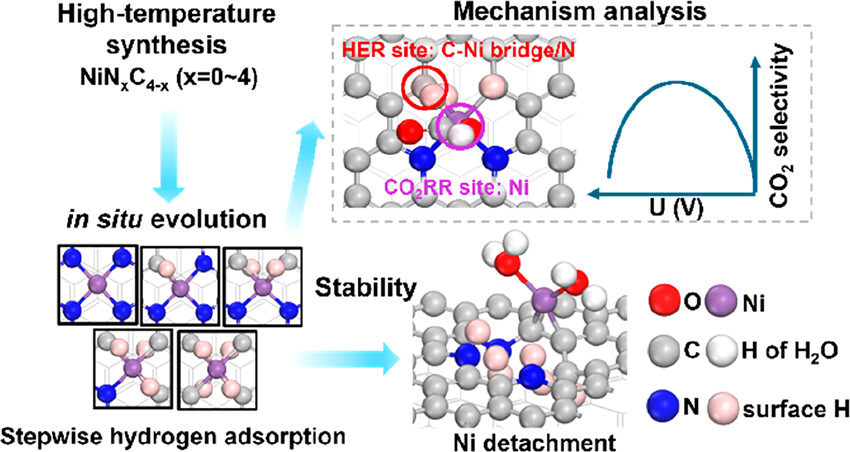
Key Points of This Article


This article develops a comprehensive theoretical framework that incorporates operational conditions, combining grand canonical ensemble density functional theory (GCDFT) and machine learning-accelerated sampling methods to reveal the structure-activity-stability relationships of Ni−N−C single-atom catalysts in the CO₂ reduction reaction. Using NiNxC4-x (x = 0−4) as a model system, representing possible coordination defects formed during high-temperature synthesis, the behavior of these sites under working conditions is systematically evaluated. By analyzing hydrogen adsorption, CO₂ adsorption and activation, CO₂RR and hydrogen evolution reaction (HER) pathways, and potential-induced structural instability, a comprehensive understanding of the catalytic performance of Ni−N−C catalysts under different conditions is achieved.
1. Hydrogen adsorption behavior: It is found that hydrogen prefers to adsorb at the C−Ni bridge sites or nitrogen atoms rather than directly on the nickel center. Hydrogen adsorption alters the coordination environment and surface structure of Ni, significantly impacting the activity and stability of the catalyst.
2. CO₂ adsorption and activation: On the unhydrogenated surface, the adsorption strength of CO₂ increases with the number of carbon atoms coordinated to nickel. However, after hydrogenation, the adsorption of CO₂ is generally weakened, and hydrogen adsorption in configurations below the graphene plane is more favorable for CO₂ activation.
3. Activity and selectivity of CO₂RR and HER: On the unhydrogenated surface, NiN3C1 exhibits the highest CO₂RR activity, while NiN₄ shows lower activity for HER. After hydrogenation, the activity of CO₂RR changes slightly, while HER activity significantly decreases due to hydrogen coverage. High CO₂RR selectivity of Ni−N−C catalysts is achieved by spatially separating active sites and suppressing HER under moderate hydrogen coverage.
4. Potential-dependent evolution of activity and selectivity: As the potential becomes more negative, the relative contributions of different hydrogenated isomers change, leading to volcano-type trends in the activity and selectivity of CO₂RR and HER. At more negative potentials, the selectivity of CO₂RR decreases, partly due to the transfer of the rate-determining process (RDP) and partly due to the co-adsorption of H and H₂O inducing the displacement of Ni atoms from the surface.
5. Stability analysis: By considering the co-adsorption of H and H₂O, it is found that the displacement of Ni atoms from the surface becomes more favorable at more negative potentials, which may partly explain the decrease in CO₂RR selectivity at more negative potentials.
Graphical Analysis


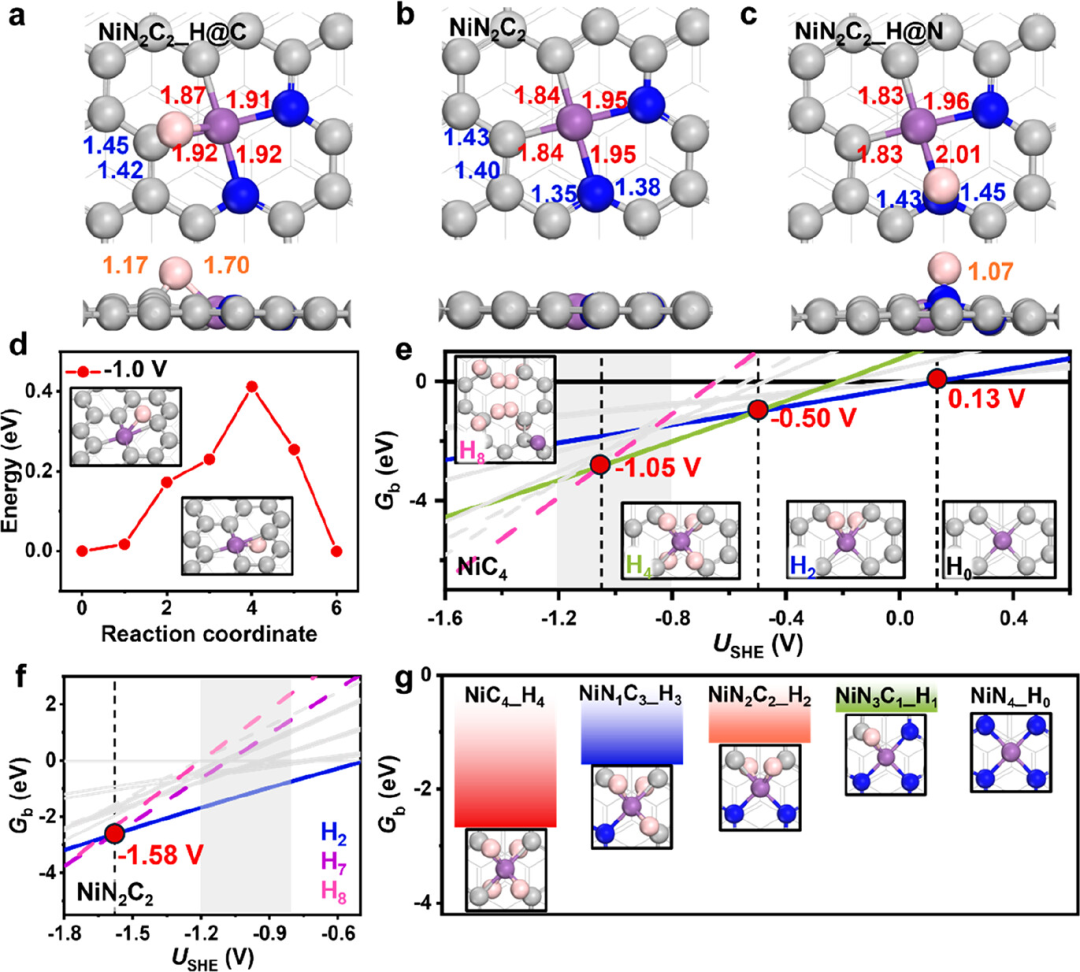
Figure 1:(a)-(c) Top and side views of the NiN2C2 configurations under different hydrogen adsorption scenarios: (a) Hydrogen (H) adsorbed at the carbon-nickel (C−Ni) bridge site; (b) pristine surface (no hydrogen adsorption); (c) Hydrogen adsorbed at adjacent nitrogen (N) atoms. Bond lengths are in angstroms (Å). (d) Energy curve for hydrogen migrating from the top to the bottom of NiC4 under conditions of -1.0 V relative to the standard hydrogen electrode (SHE). (e) Thermodynamic stability of NiC4 under different applied potentials for each coverage, considering only isomers with energies within 0.2 eV of the global minimum at -1.0 V. (f) Thermodynamic stability of NiN2C2 under different hydrogen coverages as a function of applied voltage. (g) Gibbs binding energies of the most stable hydrogen coverage configurations of different Ni−N−C motifs under conditions of -1.0 V relative to SHE.
(Image source: J. Am. Chem. Soc.)
Figure 2:(a) Gibbs free energy difference (ΔGᵦ(CO2)) between bent and linear adsorption configurations of carbon dioxide (CO2) in Ni−N−C motifs with different hydrogen coverages at -1.0 V relative to SHE. (b-c) Energy spans (Espan) analysis of CO₂ reduction reaction (CO₂RR) and hydrogen evolution reaction (HER) for different Ni−N−C motifs at -1.0 V relative to SHE. (d) Comparison of energy spans (Espan) for HER at the C−Ni bridge/N site and Ni site in the most thermodynamically stable hydrogen coverage configuration of Ni−N−C at -1.0 V. (e) Top view structure corresponding to the HER configurations analyzed in (d), with hydrogen adsorption sites highlighted in orange. (f) Schematic of spatial separation of active sites on Ni−N−C motifs: CO₂ reduction occurs at the nickel (Ni) center, while HER occurs at the C−Ni bridge or nitrogen (N) sites.
(Image source: J. Am. Chem. Soc.)
Figure 3:(a) Boltzmann populations (pi) of various hydrogen coverage isomers of NiN1C3 at different applied potentials. (b-c) Energy spans (Espan) for CO₂ reduction reaction (CO₂RR) (b) and hydrogen evolution reaction (HER) (c) of various hydrogen coverage isomers of NiN1C3 at different applied potentials. (d) Boltzmann-weighted energy spans (Espanweighted) for CO₂RR of various Ni−N−C motifs at different applied potentials. (e-f) Relative contributions of various hydrogen coverage isomers of NiN1C3 to overall CO₂RR (e) and HER (f) at different potentials. (g) Difference in Boltzmann-weighted energy spans for CO₂RR and HER of various Ni−N−C motifs at different applied potentials.
(Image source: J. Am. Chem. Soc.)
Figure 4:(a) Top view of NiN2C2 configurations with different numbers of adsorbed water molecules (1 H2O, 2 H2O, 3 H2O, and 4 H2O). (b) Gibbs binding energies of hydrogen (H) and water molecules (H2O) on NiN1C3 under conditions of -1.4 V relative to SHE. (c) Thermodynamic stability of various hydrogen (H)/water (H2O) co-adsorption configurations on NiN1C3, NiN2C2, and NiN3C1 as a function of applied potential. (d) Relationship between the initial voltage for Ni atoms to detach from the surface and the number of coordinated nitrogen (N) atoms under conditions of co-adsorption of only hydrogen (H) and hydrogen (H)/water (H2O).
(Image source: J. Am. Chem. Soc.)
Summary and Outlook


This study investigates the dynamic behavior of active sites in Ni−N−C single-atom catalysts during the CO₂ electroreduction reaction (CO₂RR) by combining grand canonical ensemble density functional theory (GCDFT) and machine learning-accelerated sampling. The study finds that under working conditions, the surface of Ni−N−C catalysts undergoes hydrogenation, with hydrogen preferentially adsorbing at C−Ni bridge sites or nitrogen atoms rather than the nickel center. This hydrogenation phenomenon significantly impacts the activity, selectivity, and stability of the catalyst. The study reveals that NiN3C1_H1 is the optimal active site, exhibiting high CO₂RR selectivity under moderate hydrogen coverage, while spatially separating active sites suppresses the hydrogen evolution reaction (HER). However, at more negative potentials, the selectivity and activity of CO₂RR decrease due to the transfer of the rate-determining process and the displacement of Ni atoms from the surface. This research emphasizes the importance of considering the coordination environment, hydrogenation state, and potential-induced structural evolution when designing the next generation of single-atom catalysts.
Reference Information


Uncovering the True Active Sites in Ni−N−C Catalysts for CO2 Electroreduction. J. Am. Chem. Soc.2025.
DOI:10.1021/jacs.5c12847
https://doi.org/10.1021/jacs.5c12847
Click to read the original article directly~
Disclaimer: For more content, please refer to the original article. If there is any infringement, please contact the editor for deletion.