After nearly 30 years of development, the ADC platform has been established, with 12 approved products by the FDA and many more in development. This article aims to provide guidance for clinical researchers on the effective design of clinical protocols and the lessons learned from ADCs.
01Introduction
In cancer treatment, compared to novel immunotherapy targets based on antibodies (LAG3), more ADCs with new targets have been approved in the past four years (tissue factor, Nectin 4, CD19, Trop 2, BCMA, CD79b). Despite progress, many recurring challenges still exist in the development of ADCs, slowing clinical advancement.
In light of this, this review aims to help clinical and industry researchers draw lessons from past successes and failures (sometimes both). Real-world examples will be used, and practical solutions will be provided to enable faster and safer development of future ADCs.
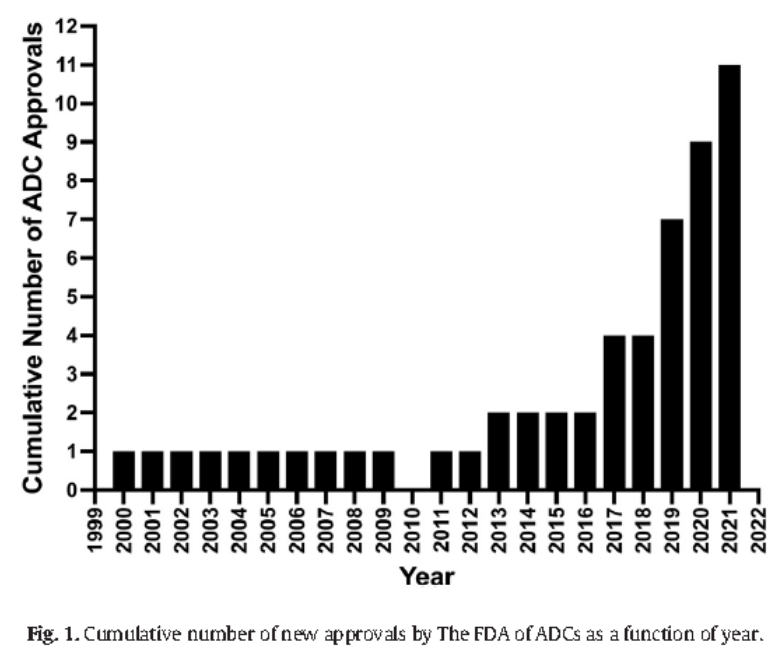
Figure 1: Cumulative number of FDA-approved ADCs
02Methods
1. Recruiting Patients from the “Real World”
Patients entering Phase I clinical trials have advanced cancer and have failed standard treatments. These patients have a higher tumor burden due to previous treatment failures and ongoing toxicity from prior therapies. Additionally, based on the demographic data of most cancer patients, many subjects have comorbidities such as hypertension, diabetes, hyperlipidemia, and venous thromboembolism, often requiring multiple medications daily. Importantly, both inclusion and exclusion criteria in Phase I oncology practice and ADC development should reflect the real patients being considered for clinical trials.
2. Inclusion and Exclusion Criteria
Inclusion criteria not only affect whether patients participate in clinical trials but also impact researchers. With so many new drugs on the market today, if the inclusion criteria are too restrictive, researchers may also refuse to participate.
It is important to note that each inclusion criterion introduces bias into the selected population and ultimately determines whether the data obtained can be generalized in future studies or even replicated. In clinical protocol writing, it is necessary to reduce “template” standards, as template standards are merely rewritten from past irrelevant studies.
3. Patient Performance Status Can Be 0-2, Not Limited to 0-1
In the United States, most patients must work to maintain health insurance, so their performance status (PS) is 0-1. Most Phase I clinical studies require extensive sampling on Day 1, followed by a series of pharmacokinetic data collection daily or weekly. Many specified and ancillary tests for ADC development, including baseline and cardiac assessments (e.g., MUGA, Echo, ECG), and ophthalmic examinations, impose a significant burden on patients.
In fact, patients with a PS of 2 are excellent candidates for studies because they have left full-time work or are exempted from work due to disability. They can successfully participate in newer ADC Phase I clinical studies through multiple visits and study authorization processes.
4. Allow Patients with Organ Abnormalities to Be Recruited
If preclinical toxicology does not show liver or kidney toxicity, the study protocol should allow for a certain degree of functional impairment in these organs. We usually recommend serum creatinine levels (ULN) up to 1.5 times the normal upper limit (including 1.5 times), and liver transaminases up to 3 times ULN (without liver metastasis). For patients with liver metastasis, we recommend a threshold of 5 times ULN.
Do not exclude patients with elevated alkaline phosphatase because alkaline phosphatase may originate from bone or liver, and if bilirubin and liver transaminases are acceptable, there is virtually no practical impact on tolerance. It is unnecessary to exclude prostate cancer, lung cancer, and breast cancer patients since these diseases have high rates of bone metastasis, leading to elevated alkaline phosphatase.
Do not exclude patients based on serum albumin levels, as there is no evidence that albumin levels of 2.8 versus 3.0 are more meaningful in clinical studies. Since albumin levels are negatively correlated with tumor burden, you will inadvertently select patients with lower tumor burdens. This restriction will lead to significant bias. If the goal is to develop drugs that can meet the unmet needs of patients (as defined by expedited approval populations), then in later studies, the response rates may decline, and you will subsequently analyze the reasons for the failed studies.
Most ADCs do not have QTc liability. Therefore, strict QTc restrictions will exclude comorbid patients for whom QTc cannot be assessed, including those with bundle branch block or pacemakers.
5. Avoid Arbitrary/Special Criteria Without Clinical Scientific Basis
Allow the use of anticoagulants and a certain degree of coagulation parameter abnormalities. Thromboembolic events are common in many cancer indications, and the use of factor Xa inhibitors can effectively control and prevent thromboembolic events. Unless thrombocytopenia is predicted to be a specific toxicity related to the ADC, restricting patients to those without anticoagulants or requiring normal coagulation parameters will exclude other excellent candidates.
It is common for advanced cancer patients to experience absolute lymphocyte count (ALC) declines as a result of disease state and prior treatments. Restricting patients with low ALC from entering studies typically offers no benefit, as this is not a predictor of toxicity for most patients. The most compelling evidence against restricting ALC comes from the initial clinical trials of pembrolizumab. ALC standards should not be included in ADC development protocols.
6. Do Not Exclude Patients Who Have Previously Received ADC Treatment
Excluding patients who have been exposed to previous ADCs can reduce the risk of developing resistance to the latest ADC. However, the reasons for a patient’s lack of response to ADCs are multifactorial, including receiving subtherapeutic dose levels, low target antigen expression levels in the tumor, unstable linkers, or inherent resistance of the tumor to the payload.
The strongest possible argument against excluding patients with prior ADC treatment is that new therapies must meet the unmet needs of patients, and the fastest route to approval is based on the benefits seen in patients who have failed previous treatments. The accelerated approval of fam-trastuzumab deruxtecan (Enhertu®) for HER2-positive metastatic breast cancer is a great example. This study specifically included patients with HER2-positive breast cancer who had previously failed ado-trastuzumab emtansine (Kadcyla®). The significant response rate and prolonged response time accelerated the approval process. If the study excluded prior ADC treatment, it would be difficult to determine the currently available ADC treatments (ado-trastuzumab emtansine), and it would likely lead to traditional approval based on comparisons with Kadcyla®.
All inclusion criteria for protocols should learn from past experiences.Pembrolizumab Although it is an immunotherapy, it had almost no restrictive inclusion criteria in its early Phase I development, and the drug completed its dose-finding phase in 8 months, from the first patient in Phase I clinical trials to FDA approval time of 3.4 years.
03Results
MTD is usually derived from the safety data of the first cycle. However, in practice, drugs are used over many cycles. Only after several cycles do cumulative toxicities become apparent, some of which become problematic. Some promising ADCs are associated with life-threatening pneumonia, which only occurs after multiple cycles. Therefore, the recommended dose should be determined after carefully analyzing multiple patients, multiple cycles, and the number of dose reductions and delays. Spending more time in Phase I clinical trials to adjust the recommended dose is much better than announcing that the dose is being reduced in the registration study.
We have ample evidence that after the approval of capecitabine and regorafenib, physicians did not routinely prescribe the approved full doses.
The therapeutic index of ADCs is narrower than that of non-ADC antibody therapies, and careful analysis of the relationship between dose, administration formulation, clinical pharmacokinetics, and toxicity is required to determine the optimal administration method (see Figure 2).
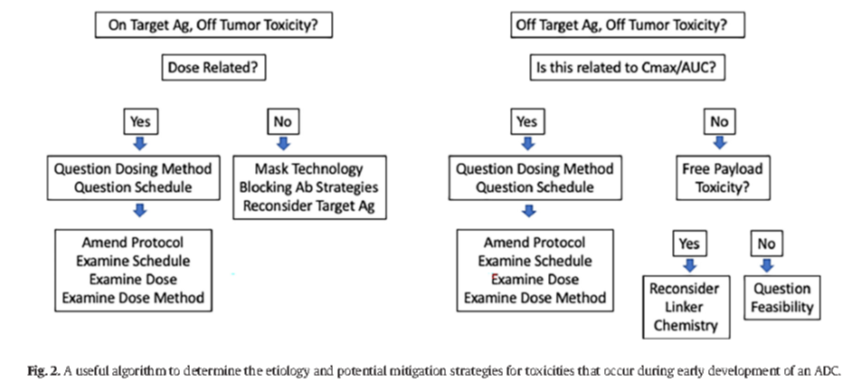
Figure 2: Useful algorithm for identifying the causes of toxicity occurring during the early development of ADCs and potential mitigation strategies
04Exploring Diverse Administration Cycles
Cytotoxic drugs are typically chosen to be administered every 3 weeks, as this closely aligns with hematologic recovery time.
Antibody drugs are increasingly administered based on expected clearance and elimination half-lives, typically every 2 to 4 weeks. For antibody-drug conjugates, there is no need to adopt the schedules of cytotoxic or antibody eras.A better option is to select administration cycles based on tolerance, pharmacokinetics, and/or target antigen turnover and regeneration kinetics.
05Strategies for Antigen Biomarkers
The primary strategy for developing ADC biomarkers is to identify and select cancer patient subgroups most likely to respond to ADCs, thereby achieving high rates and durable responses and ensuring regulatory approval, which may be the biggest challenge in biomarker development. To guide how to proceed, several practical recommendations are listed below.
First is the involvement of experts in the companion diagnostics field.
Second is early recruitment of patients who are most likely to express the antigen.
Third is parallel development of clinical trials and biomarkers, emphasizing the need to avoid prospective selection of patients in early clinical trials and allowing optimization of testing methods and retrospective analyses to guide patient and method selection.
Not all ADCs require biomarkers for patient selection. If biomarkers eliminate potentially beneficial treatments for patients, it may limit future commercial interests.
Finally, in registration studies, do not arbitrarily change truncation parameters. One instance is mirvetuximab soravtansine, an ADC targeting folate receptor alpha (FRa) for the indication of refractory ovarian cancer. The Phase II study observed antitumor activity in patients with higher expression of FRa. Unfortunately, the analysis of the registration study (Forward 1) was changed to include patients with intermediate expression of FRa, leading to not meeting the primary endpoint of progression-free survival (PFS) improvement. Post hoc analysis identified a high-expression (FRa) patient group that achieved significant statistical significance, similar to the patients selected in early studies. Further attempts are using analysis truncations very similar to the original patient selection criteria.
06Clinical Pharmacology Considerations for ADCs
Many of the clinical pharmacology steps required for regulatory approval in ADC development are not listed separately. From the perspective of the drug or biologic, should ADCs be viewed as cytotoxic drugs or closer to antibodies? This uncertainty leads to questions about whether organ function impairment studies, drug interaction studies, and QTc studies are needed. The FDA recently released draft guidance to assist in the clinical development of ADCs. Very practical recommendations assist researchers in determining clinical pharmacology. It can also be argued that this guidance further proves that ADCs themselves represent a platform that transcends cytotoxic drugs and antibody drugs.
References
Anthony W. Tolcher, Pharmacology & Therapeutics 240 (2022) 108235, https://doi.org/10.1016/j.pharmthera.2022.108235
Editor: 💧Transparent This tweet is used to disseminate knowledge. If you have any questions regarding copyright, please contact us within 30 days of this article’s publication.Original content is prohibited from being reproduced on other platforms without authorization.If you have questions, please send an email to [email protected] for more information.©2021 Medical Overview All rights reservedPrevious Links“The Journey of the Little Vaccine” | Pharmaceutical Company Pipeline Review Everyone Understands Immunology | Everyone Understands Immunology (Audio Version) Review Article Interpretation | Literature Brief | Medical Popular Science | Pharmaceutical Frontier NotesPROTAC Technology | Antibody Drugs | Antibody-Drug Conjugates – ADCNucleic Acid Vaccines | CAR Technology | Chemical Biology


Warm Reminder
 The Medical Overview WeChat public account currently has nearly 12 discussion groups (enthusiastic, interesting, and gathering talents in the pharmaceutical circle). To join the group, add the author’s WeChat (yiyaoxueshu666) or scan the QR code of the public account to add the author, and note “Name/Nickname – Company/University – Specific Research Field/Major”. This group is only for scientific research discussions, please do not disturb.Simple operations to star⭐️ Medical Overview, receive our push notifications first time① Click on “Medical Overview” below the title ② Go to the top right corner “…” ③ Click “Set as Star”
The Medical Overview WeChat public account currently has nearly 12 discussion groups (enthusiastic, interesting, and gathering talents in the pharmaceutical circle). To join the group, add the author’s WeChat (yiyaoxueshu666) or scan the QR code of the public account to add the author, and note “Name/Nickname – Company/University – Specific Research Field/Major”. This group is only for scientific research discussions, please do not disturb.Simple operations to star⭐️ Medical Overview, receive our push notifications first time① Click on “Medical Overview” below the title ② Go to the top right corner “…” ③ Click “Set as Star”