Click the blue text

Follow us
This
Issue

Focus
Point
B7-H4 is a member of the B7 immune checkpoint ligand family, which has been shown to bind to an unknown receptor on T cells, inhibiting T cell proliferation and cytokine production, thereby negatively regulating T cell function.
The expression level of B7-H4 is elevated in various solid tumors, including ovarian cancer, breast cancer, endometrial cancer, cholangiocarcinoma, gallbladder cancer, and squamous non-small cell lung cancer. In normal tissues, B7-H4 is only expressed at low levels in immune cells, making it an ideal molecular target for ADCs. However, among such a popular target, some companies rejoice while others face disappointment. Pfizer has terminated its B7H4 ADC project, while Hansoh has already initiated Phase III clinical trials. BeiGene has introduced the B7H4 ADC from I-Mab for over $1.3 billion, attempting to achieve a breakthrough by combining it with Tislelizumab. At the recently concluded 2025 ASCO conference, the reported data for two B7H4 ADCs fell short of expectations, raising concerns about the drug development risks associated with this target. This article will systematically review and present the current status and outlook of this field.
Content of this issue
|
01 |
2025 ASCO | B7H4 ADC Data Disclosure |
|
02 |
Pfizer | BeiGene | I-Mab |
|
03 |
Hansoh Pharmaceutical: Advancing to Phase III, Accelerating Breakthrough Therapy Designation |
|
04 |
Summary and Outlook |
【01 2025 ASCO | B7H4 ADC Data Disclosure】
BeiGene Partners with I-Mab for Over $1.3 Billion
On May 7, 2025, BeiGene received clinical implied approval from the CDE for the B7-H4 ADC drug BG-C9074, developed in collaboration with I-Mab, to be combined with the PD-1 monoclonal antibody Tislelizumab for the treatment of advanced solid tumors. This marks the second clinical advancement for the drug following its approval for monotherapy indications in July 2024, indicating a comprehensive exploration from monotherapy to combination therapy.
Global Layout: BG-C9074 (I-Mab’s development code DB-1312) was licensed to BeiGene in July 2023 for over $1.3 billion and has initiated Phase I clinical trials in the US and Australia, planning to enroll 150 patients.
Domestic Progress: In July 2024, BeiGene initiated Phase Ia/Ib trials in China, focusing on safety and anti-tumor activity to pave the way for subsequent registration.
At the 2025 ASCO conference, preliminary results from the first human study of BG-C9074 in patients with advanced solid tumors were disclosed during the dose escalation phase. Let’s take a look at the specific research data.

Methods:
BG-C9074-101 (NCT06233942) is a first-in-human, multicenter study aimed at evaluating the safety, tolerability, pharmacokinetics, and preliminary anti-tumor activity of BG-C9074 as monotherapy and in combination with Tislelizumab in patients with advanced solid tumors (according to RECIST v1.1 criteria). The study included patients with histologically or cytologically confirmed locally advanced, unresectable, or metastatic solid tumors, regardless of B7-H4 expression levels. Patients received BG-C9074 intravenously every three weeks, with dose cohorts escalating from 1 mg/kg to 7 mg/kg.
Results:
As of January 22, 2025, a total of 55 patients with advanced solid tumors received BG-C9074 monotherapy (including 25 with ovarian cancer, 16 with breast cancer, 10 with cholangiocarcinoma, and 4 with other tumor types). 3 patients experienced dose-limiting toxicities (fatigue in the 6 mg/kg group, febrile neutropenia and thrombocytopenia in the 7 mg/kg group). 48 patients (87.3%) reported treatment-related adverse events (TEAEs), with 27.3% of patients experiencing ≥ grade 3 TEAEs. The most common TEAEs were nausea (45.5%), fatigue (38.2%), and neutropenia (32.7%), with neutropenia being the most common ≥ grade 3 TEAE (16.4%). Among 39 patients with evaluable efficacy, 8 (20.5%) achieved partial responses (4 confirmed, 4 unconfirmed).
Conclusion:
BG-C9074 demonstrated controllable safety and tolerability in patients with advanced solid tumors expressing B7-H4. Preliminary clinical responses were observed across various dose levels and tumor types, without selective screening for B7-H4 expression. Dose escalation and expansion studies are ongoing.
Emi-Le is a B7-H4 ADC developed by Mersana Therapeutics, with the development code XMT-1660. This drug utilizes Mersana’s proprietary Dolasynthen technology platform, combining DolaLock microtubule inhibitor payload design to achieve targeted killing of tumor cells while limiting bystander effects. XMT-1660, as one of its core pipelines, initiated Phase I clinical trials in 2023, focusing on dose escalation studies in patients with breast cancer, endometrial cancer, and ovarian cancer. Clinical data released in January 2025 showed that the drug exhibited preliminary efficacy in patients with high B7-H4 expression in triple-negative breast cancer, with an ORR of 23%, but limited efficacy in low-expression patients.
Preliminary data from the Phase I dose escalation study of Emi-Le was presented at 2025 ASCO.
Methods:
This Phase I study evaluates the safety and preliminary anti-tumor activity of Emi-Le monotherapy in adult patients with advanced/metastatic triple-negative breast cancer (TNBC), HR⁺/HER2⁻ breast cancer, OC, EC, and ACC-1. During the dose escalation phase, eligible patients received Emi-Le at doses ranging from 7.2 to 115 mg/m² per cycle, with all safety and efficacy data used to determine the recommended dose for the expansion phase (EXP). The study conducted retrospective B7-H4 expression immunohistochemistry (IHC) testing on tumor specimens, with preliminary high expression cutoff set at TPS ≥ 70.
Results: As of December 13, 2024, a total of 130 patients received treatment. The median age of all patients was 55 years, with a history of 4.5 lines of systemic therapy (range 0–15 lines). 103 patients were assessed for B7-H4 status, of which 44% were high expressers (high TPS). Overall, Emi-Le was well tolerated. The most common treatment-related adverse events (TRAEs) were transient AST elevation (38%, 14% grade 3), proteinuria (31%, 9% grade 3), nausea (29%, 1% grade 3), and fatigue (28%, no ≥ grade 3). The only grade 3 TRAEs occurring in ≥5% of patients were AST elevation and proteinuria, with no grade 4 or 5 TRAEs reported. No dose-limiting neutropenia, neuropathy, ocular toxicity, interstitial lung disease, or thrombocytopenia were observed. 2.3% of patients discontinued treatment due to TRAEs. Clinical activity correlated with dose and B7-H4 expression levels. In the mid-dose range (38.1–67.4 mg/m²/cycle), among evaluable high expressers, the confirmed ORR was 23% (6/26); TNBC patients (13 patients, all previously received at least one line of topoisomerase I ADC) had a confirmed ORR of 23% (3/13). In the high-dose range (≥76.2 mg/m²/cycle), high expressers had a confirmed ORR of 22% (2/9), with 78% (7/9) of patients showing target lesion shrinkage ≥30%. Among the 8 patients with confirmed responses at doses ≥38.1 mg/m²/cycle, 5 had target lesions shrinkage >60%, including 1 complete response. All 4 high expressers receiving the initial EXP dose of 67.4 mg/m² every four weeks showed tumor shrinkage, with treatment ongoing for ≥16 weeks as of the data cutoff.
Conclusion: Based on preliminary data, Emi-Le demonstrated encouraging clinical activity and good tolerability in heavily pre-treated patients. The EXP study at a dose of 67.4 mg/m² Q4W is currently ongoing, enrolling patients with advanced/metastatic TNBC who have previously received 1–4 lines of systemic therapy; a second higher EXP dose is also being explored. Mersana currently faces pipeline reduction challenges, with early ADC projects like XMT-1522, XMT-1592, and XMT-2056 either terminated due to safety issues or placed on clinical hold. XMT-1660, as one of its remaining core projects, carries significant expectations for the company’s future development.
However, from the above two studies, as well as previous early data from AstraZeneca’s B7H4 ADC (AZD8205), the ORR in solid tumor patients has not exceeded 25%. Blindly advancing to Phase II/III indeed poses risks, and Pfizer has already terminated the development of the B7H4 ADC project.

02 Pfizer Terminates B7H4 ADC Project
Pfizer completed its acquisition of Seagen in 2023, obtaining the B7H4 ADC drug Felmetatug vedotin.
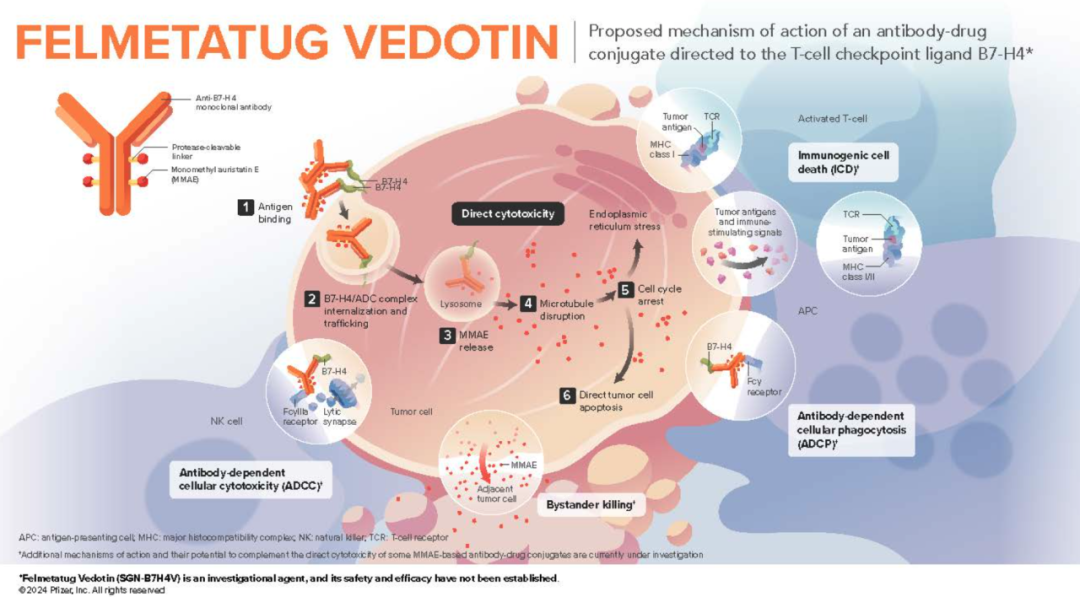
Mechanism of Action of Felmetatug vedotin
Felmetatug vedotin consists of three components: a monoclonal antibody targeting B7-H4, a microtubule disruptor MMAE, and a protease-cleavable mc-vc linker that covalently connects MMAE to the antibody, allowing MMAE to be preferentially released within target cells.
However, due to the latest clinical data indicating that Felmetatug vedotin is unlikely to achieve significant improvement over standard chemotherapy in patients with advanced solid tumors, Pfizer has decided to terminate the development of this drug and has recorded a $1 billion impairment of intangible assets.
Although Pfizer has terminated the B7H4 ADC project, research on the B7H4 target continues. Research in the B7H4 ADC field may increasingly focus on biomarker strategies to identify patient populations most likely to benefit from treatment.
In summary, despite Pfizer’s setbacks with the B7H4 ADC project, other companies like Hansoh Pharmaceutical are still actively advancing related research. The global ADC market is rapidly growing, and the research and application prospects for the B7H4 target remain broad.
【03 Hansoh Pharmaceutical: Advancing to Phase III, Accelerating Breakthrough Therapy Designation】
Hansoh Pharmaceutical’s HS-20089 (GSK5733584) has become the focus of the field as the “first B7-H4 ADC to enter Phase III clinical trials globally”:
Impressive Clinical Data:
Phase I data presented at ESMO 2023 showed that HS-20089 achieved an objective response rate (ORR) of 41.7% in patients with triple-negative breast cancer, with patients in the lowest dose group maintaining responses for over 403 days. In March 2025, its Phase III clinical trial targeting platinum-resistant ovarian cancer was initiated, directly competing with standard chemotherapy regimens and potentially filling a gap in this field.
Accelerated International Collaboration:
In 2023, Hansoh reached a $1.57 billion licensing agreement with GSK, with GSK leading overseas development. In April 2025, HS-20089 received breakthrough therapy designation from the NMPA, which is expected to accelerate its market entry.
04 Summary and Outlook
With innovations in ADC technology and clinical strategies from Chinese pharmaceutical companies, the B7-H4 target is expected to become the next “super track” following PD-1. The leadership of BeiGene and Hansoh not only promises more precise treatment options for patients but also marks a leap from “following” to “leading” in Chinese innovative drugs. In the future, combination therapies and precise biomarker screening may become core breakthroughs, potentially ushering in a new chapter in global cancer treatment defined by China.
★
