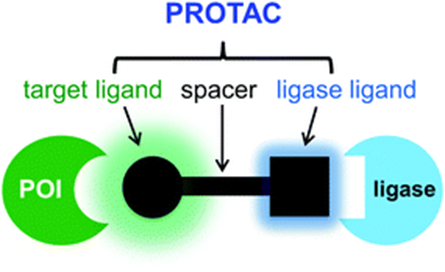
Protein Hydrolysis Targeted Chimeras (PROTAC) are bringing revolutionary changes to the fields of biology and medicinal chemistry. PROTAC consists of a target protein ligand, an E3 ligase ligand, and a spacer group[Unlike the linker used in antibody-drug conjugates, here we specifically use “spacer” instead of the commonly seen “linker” in PROTAC literature].
PROTAC structural composition (Source: Chemical Society Reviews)
As antibody-drug conjugates (ADCs) continue to demonstrate clinical feasibility and commercial value, many research groups have attempted to mimic the design principles of ADCs by linking PROTACs to monoclonal antibodies (mAbs) to enhance the in vivo delivery efficiency of PROTACs. This form of medication, called Degrader-Antibody Conjugates (DACs), can overcome the following issues of unconjugated PROTAC molecules: (1) low in vivo delivery efficiency due to physicochemical properties and DMPK properties (especially when E3 ligase is not CRBN); (2) complex formulations used to achieve sufficient exposure; (3) non-specific targeting of PROTAC.
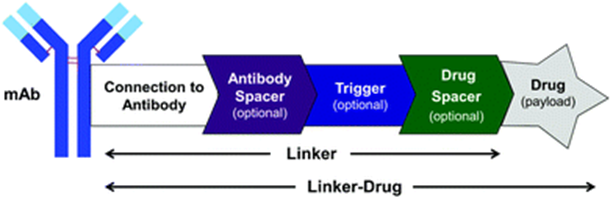
ADCs combine cytotoxic small molecule drugs (payloads) with mAbs through linker groups. To enhance the in vivo activity of ADCs, the structure may sometimes include (1) a spacer element that separates the linker group from the antibody (Antibody Spacer); (2) a chemical or biological trigger element that activates intracellular cleavage (Trigger); (3) a second spacer element that separates the trigger element from the small molecule drug (Drug Spacer).(Note: Different colored blocks in the figure correspond to modules in subsequent illustrations)
ADC structural composition (Source: Chemical Society Reviews)
Small molecule drugs are conjugated to linker groups through chemical synthesis and then linked to the surface of the antibody through conjugation techniques. Drugs attached to the surface of the antibody cannot bind to the target. Once the antibody in the ADC structure binds to the cell surface antigen, the entire ADC molecule is internalized. The antigen targeted by the ADC must be highly expressed in the target tissue, quickly internalize the ADC, and have appropriate transport capabilities, while being expressed at relatively low levels in non-target tissues.
The internalized ADC molecule is then transported to the lysosome, where the small molecule drug is released. Various lysosome-responsive cleavage methods have been developed, such as acid hydrolysis, disulfide bond reduction, and peptidase cleavage. ADCs with different cleavage methods can remain stable in circulation but release the small molecule only in the lysosome of the target cells. The small molecule is actively transported out of the lysosome, diffusing into the cytoplasm, and subsequently interacting with the target. This requires that the small molecule drug must be very stable in the lysosome to successfully interact with the target.
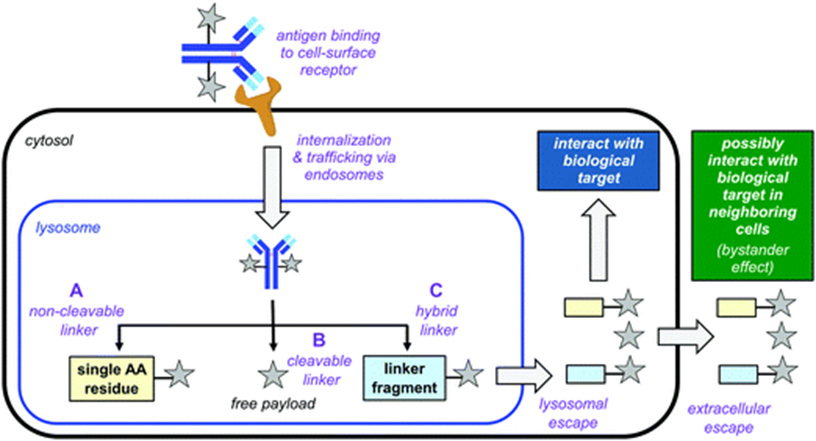 Mechanism of action of ADCs. (A) Non-cleavable linker: The antibody is metabolized by lysosomal enzymes, and the drug binds to the linker and a small part of the antibody structure (usually a single amino acid); (B) Cleavable linker: The trigger element activates cleavage in the lysosome, releasing free small molecules; (C) In some cases, the small molecule may bind to fragments of the linker after cleavage (Source: Chemical Society Reviews)
In some cases, the released small molecule drugs may enter adjacent cells, which may or may not express the target antigen. This “bystander effect” can kill related cells that do not express the target antigen (alleviating the tumor heterogeneity issue), but it may also cause off-target toxicity to non-target cells.
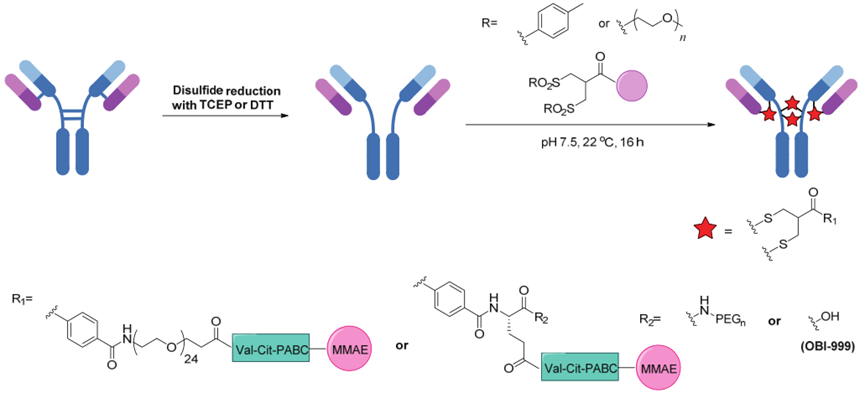
Currently, the conjugation technology for ADC drugs has made significant advancements. The first category of technology involves using N-hydroxysuccinimide (NHS) esters to activate carboxylic acids for coupling with lysine residues on antibodies. The first approved ADC drug, Mylotarg, was achieved using this technology; however, this method produces a wide range of drug-antibody ratios (DAR, i.e., the number of drug molecules conjugated to a single mAb) ranging from 2 to 7. The second category of technology involves using thiol-specific reagents (such as maleimide) to react with cysteine residues.Using the maleimide method, ADC molecules can form DAR of 4 (partial reduction, lower uniformity) or 8 (complete reduction, higher uniformity).
The disulfide bond bridging method involves reducing the four pairs of interchain disulfide bonds in the antibody using reducing compounds[such as tris(2-carboxyethyl)phosphine (TCEP)] and then re-bridging the disulfide bonds in the antibody using reagents that selectively react with cysteine residues while introducing cytotoxic drugs. This method not only maintains the stability of the original disulfide bonds but also controls that only one linker molecule is conjugated to each pair of disulfide bonds, achieving site-specific conjugation.
Using bisulfide compounds to re-bridge interchain disulfide bonds in antibodies produces homogeneous ADCs (Source: Nature)
DAR requirements are related to the activity of small molecule drugs; the stronger the small molecule activity, the lower the required DAR. Some small molecules in ADCs inhibit microtubule protein polymerization, disrupting microtubule activity, or directly kill DNA through alkylation, or inhibit DNA topoisomerase 1 activity to kill tumors. Common small molecules in ADCs exhibit strong cytotoxic characteristics in vitro, with anti-proliferation IC50 reaching sub-nanomolar levels, requiring only 2-4 DAR to be effective. However, the activity of DNA topoisomerase 1 inhibitors is slightly lower, requiring 4-8 DAR in ADCs containing DNA topoisomerase 1 inhibitors to trigger sufficient cell-killing activity. However, as DAR increases, ADCs tend to aggregate, which is detrimental to tumor-killing effects.
Mechanism of action of ADCs. (A) Non-cleavable linker: The antibody is metabolized by lysosomal enzymes, and the drug binds to the linker and a small part of the antibody structure (usually a single amino acid); (B) Cleavable linker: The trigger element activates cleavage in the lysosome, releasing free small molecules; (C) In some cases, the small molecule may bind to fragments of the linker after cleavage (Source: Chemical Society Reviews)
In some cases, the released small molecule drugs may enter adjacent cells, which may or may not express the target antigen. This “bystander effect” can kill related cells that do not express the target antigen (alleviating the tumor heterogeneity issue), but it may also cause off-target toxicity to non-target cells.
Currently, the conjugation technology for ADC drugs has made significant advancements. The first category of technology involves using N-hydroxysuccinimide (NHS) esters to activate carboxylic acids for coupling with lysine residues on antibodies. The first approved ADC drug, Mylotarg, was achieved using this technology; however, this method produces a wide range of drug-antibody ratios (DAR, i.e., the number of drug molecules conjugated to a single mAb) ranging from 2 to 7. The second category of technology involves using thiol-specific reagents (such as maleimide) to react with cysteine residues.Using the maleimide method, ADC molecules can form DAR of 4 (partial reduction, lower uniformity) or 8 (complete reduction, higher uniformity).
The disulfide bond bridging method involves reducing the four pairs of interchain disulfide bonds in the antibody using reducing compounds[such as tris(2-carboxyethyl)phosphine (TCEP)] and then re-bridging the disulfide bonds in the antibody using reagents that selectively react with cysteine residues while introducing cytotoxic drugs. This method not only maintains the stability of the original disulfide bonds but also controls that only one linker molecule is conjugated to each pair of disulfide bonds, achieving site-specific conjugation.
Using bisulfide compounds to re-bridge interchain disulfide bonds in antibodies produces homogeneous ADCs (Source: Nature)
DAR requirements are related to the activity of small molecule drugs; the stronger the small molecule activity, the lower the required DAR. Some small molecules in ADCs inhibit microtubule protein polymerization, disrupting microtubule activity, or directly kill DNA through alkylation, or inhibit DNA topoisomerase 1 activity to kill tumors. Common small molecules in ADCs exhibit strong cytotoxic characteristics in vitro, with anti-proliferation IC50 reaching sub-nanomolar levels, requiring only 2-4 DAR to be effective. However, the activity of DNA topoisomerase 1 inhibitors is slightly lower, requiring 4-8 DAR in ADCs containing DNA topoisomerase 1 inhibitors to trigger sufficient cell-killing activity. However, as DAR increases, ADCs tend to aggregate, which is detrimental to tumor-killing effects.
When developing DACs, due to the specificity of PROTAC molecules, ADC delivery strategies cannot be simply copied. Unlike the broad-spectrum cytotoxic small molecules used in ADCs, PROTACs in DACs typically only have targeted activity against specific tumors and/or tissues or cells. Therefore, the selection of antigens must not only meet the function of internalizing DAC and transporting it to the lysosome but also require high expression in the PROTAC target tissue (or tumor, cell). This antigen may also have lower levels of expression in other tissues or cells, as long as such tissues or cells have good tolerance to the PROTAC to avoid off-target toxicity.
Since the in vitro activity of PROTACs is often lower than that of small molecules, it is necessary to increase the DAR in DACs to exert efficacy (i.e., DAC > 4). However, increasing DAR may lead to DAC aggregation, adversely affecting in vivo pharmacokinetics (PK). However, compared to small molecules, PROTACs are larger and more lipophilic, making aggregation and PK issues more severe, necessitating the development of new linker groups and conjugation methods to address these problems.
Many PROTACs lack sites (amines) that can be covalently bonded to cleavable linker groups. Therefore, consideration must be given to whether it is necessary to modify the PROTAC structure to introduce reactive sites (which may alter the physiological activity of PROTAC); or to utilize existing functional groups in PROTAC (such as hydroxyl, phenolic hydroxyl) and develop new conjugation techniques. Developing non-cleavable groups also requires consideration of the same issues, ensuring that linker groups remaining attached to PROTAC after lysosomal degradation do not interfere with its biological activity.
In addition, other issues that need to be addressed in DAC development include: (1) the stability of PROTACs in lysosomes; (2) the escape function of PROTACs from lysosomes; (3) the bystander effect of PROTACs. The last two are influenced by the cell permeability of PROTACs, which remains a key focus of research in this field. PROTACs that cannot exert biological activity due to cell permeability can utilize mAbs in DACs to enter cells and exert effects, but this bystander effect of PROTACs is expected to be minimal. In this case, it is necessary to use cell-independent methods to assess the performance of PROTACs in forming ternary complexes.
Case Study: Targeting BRD4 with DAC
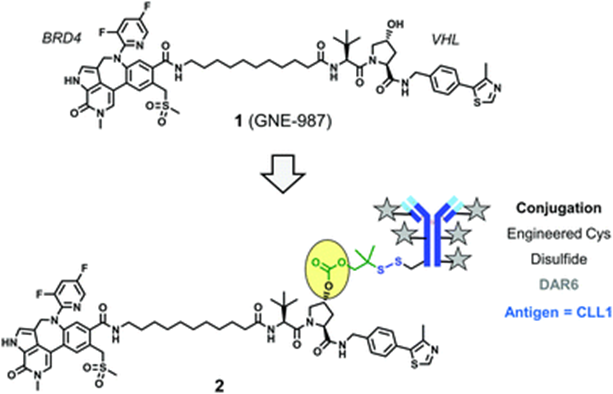
GNE-987 (Compound 1) is a PROTAC that degrades Bromodomain Protein 4 (BRD4), composed of a VHL ligand, a BRD4 ligand, and a spacer group. By using a cleavable linker group containing disulfide bonds, GNE-987 is conjugated to an antibody targeting C-type lectin-like molecule 1 (CLL1), forming a DAC (Conjugate 2). This DAC utilizes the hydroxyl of hydroxyproline in the VHL ligand structure to bind to the linker group. A single antibody’s heavy and light chains can bind six PROTAC molecules through disulfide bonds.
Conjugate 2 (Source: Chemical Society Reviews)
After a single intravenous injection, conjugate 2 showed strong, dose-dependent in vivo activity in acute myeloid leukemia (AML) HL-60 and EOL-1 xenograft models. In contrast, the activity of unconjugated compound 1 was hindered by unfavorable PK properties, resulting in poor in vivo activity. The above results indicate that DACs can overcome the suboptimal PK characteristics of PROTACs, achieving acceptable lysosomal stability and appropriate antigen targeting capabilities.
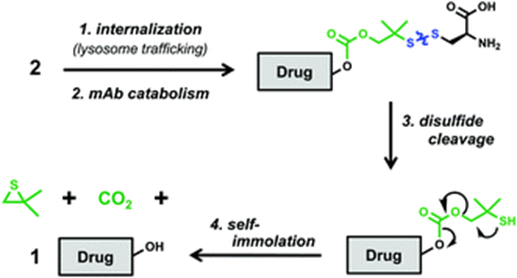
The antibody region of conjugate 2 binds to the CLL1 receptor on tumor cells, leading to internalization of the conjugate and transport to the lysosome. The antibody is hydrolyzed into amino acids in the lysosome, but a cysteine residue remains bound to the linker group through a disulfide bond. The disulfide bond is then reduced and rearranged, exposing the hydroxyl in the VHL ligand, releasing free GNE-987.
Mechanism of free PROTAC release from conjugate 2 (Source: Chemical Society Reviews)
Shortly after the announcement of compound 2, a second DAC drug targeting BRD4 emerged. In this drug, four PROTAC molecules (Compound 3) are conjugated to the surface of the antibody using interchain cysteine residues through the disulfide bond bridging method. During the conjugation process, an ester bond is formed between the hydroxyl in the VHL ligand structure and a non-cleavable linker, and finally, a cycloalkyne is formed using strain-promoted azide-alkyne cycloaddition (SPAAC) from click chemistry, resulting in a complete DAC drug (Conjugate 4). After cellular uptake, the drug in conjugate 4 completes ester bond hydrolysis in the lysosome, releasing free PROTAC molecules.
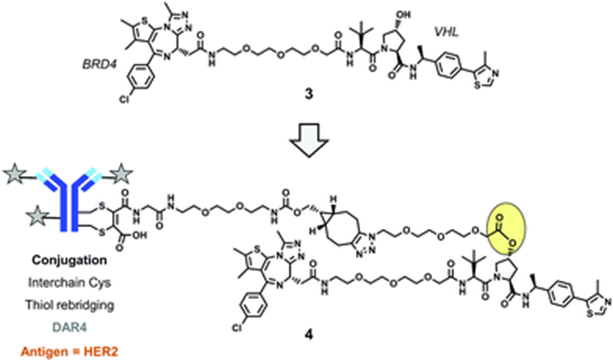
Conjugate 4 (Source: Chemical Society Reviews)
Conjugate 4 targets the human epidermal growth factor receptor 2 (HER2), which is a widely used antigen in ADCs. In vitro experiments show that conjugate 4 degrades BRD4 in HER2-positive cells in a dose-dependent manner, while no such phenomenon is observed in HER2-negative cells.
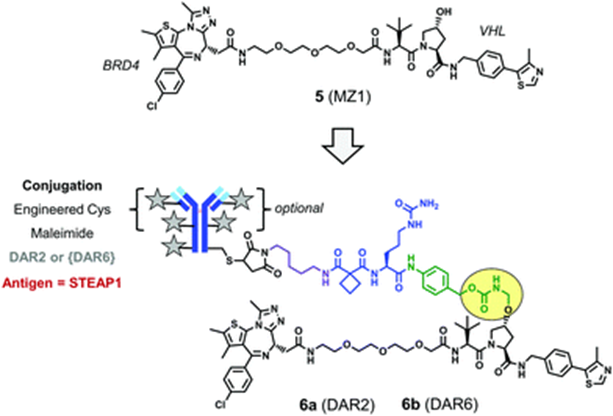
The PROTAC drug MZ1, which degrades BRD4 (Compound 5), is conjugated to an antibody using a novel linker group that contains an enzyme-cleavable peptide trigger element and a drug spacer element, formingConjugate 6. The antibody of conjugate 6 targets prostate six-transmembrane epithelial antigen 1 (STEAP1), which is highly expressed on the surface of the prostate.
Conjugate 6 (Source: Chemical Society Reviews)
In PC3 prostate cancer cells induced to express STEAP1 antigen (PC3-S1), conjugate 6b with a DAR of 6 shows stronger degradation activity than conjugate 6a with a DAR of 2. Therefore, increasing DAR can improve the biological activity of the conjugate. However, preparing higher DAR conjugates requires extensive experience from researchers, as drug molecules are influenced by multiple factors (mAb antigen, linker group structure, PROTAC lipophilicity, etc.).
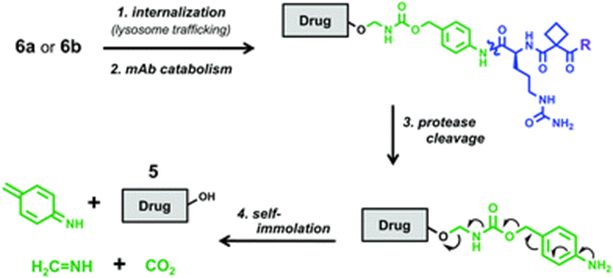
After the antibody of conjugate 6 binds to the STEAP1 receptor, the drug is internalized, transported to the cell lysosome, undergoes antibody hydrolysis, peptide cleavage, and rearrangement of the drug and spacer element, releasing free PROTAC molecules.
Cleavage pathway of conjugate 6 (Source: Chemical Society Reviews)
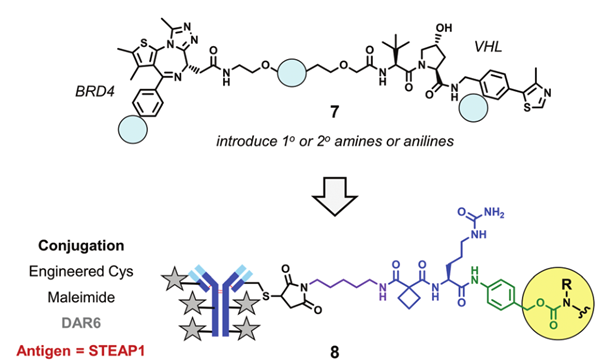
Due to the structural specificity of PROTAC drugs, it is sometimes necessary to introduce amine or aniline to form binding sites for linker groups in the PROTAC structure. For example, by introducing primary amines, secondary amines, or anilines at different positions in the structure of compound 5, the modified PROTAC molecules can be conjugated to enzyme-cleavable peptide linker groups via carbamate and then linked to the targeting STEAP1 antibody.
Experiments in PC-S1 cells found that compared to compound 5, the modified PROTAC molecules had reduced degradation ability against BRD4, possibly due to the introduction of polar functional groups disrupting the cell membrane permeability of PROTAC. Surface plasmon resonance (SPR) experiments indicated that the modified PROTAC molecules could form ternary complexes with BRD4 and VHL, with a strong correlation between the half-life of the ternary complex and BRD4 degradation activity.
DAC designed based on the structural modification of compound 5 (Source: Chemical Society Reviews)
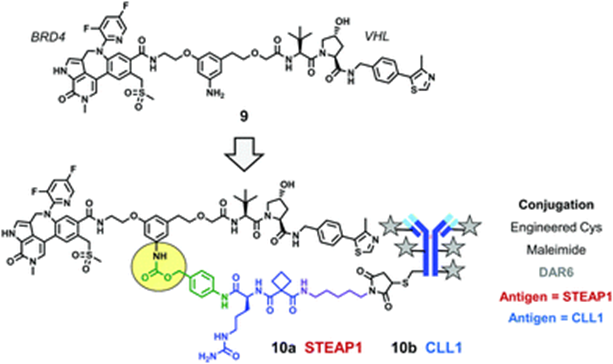
To further enhance BRD4 degradation activity and achieve stronger anti-cell proliferation activity, a series of new BRD4 degraders (Compound 9) were developed based on stronger affinity BRD4 ligands. The conjugation of compound 9 to mAbs targeting STEAP1 resulted in multiple DACs with in vitro anti-proliferative activity.
For example, compound 9 was conjugated to mAbs targeting STEAP1 or CLL1 using peptide linker groups, formingConjugate 10a andConjugate 10b. DAC 10a exhibited significant antigen-dependent anti-proliferative effects in PC3-S1 cells; DAC 10b demonstrated antigen-dependent tumor-killing effects in AML HL-60 xenograft models.
Conjugate 10 (Source: Chemical Society Reviews)
However, replacing the antibody of conjugate 2 with an mAb recognizing STEAP1, forming a conjugate with a DAR of 6, leads to DAC aggregation and loss of biological activity. Therefore, these results indicate that when developing high DAR conjugates using highly lipophilic PROTACs, it is essential to simultaneously consider the mAb antigen and the linker-drug to develop active DAC drugs.
The toxicity of conjugates 10a and 10b to megakaryocytes is reduced compared to the unconjugated compound 9, indicating that conjugating PROTACs to mAbs can improve the preclinical therapeutic index of DACs, but specific conclusions still require further experimental validation.
Recently, a new DAC drug degrading BRD4 has been reported, designed based on CRBN. The PROTAC that degrades BRD4 (Compound 11) is conjugated to a HER2 mAb through interchain cysteine residues, formingConjugate 12. In cellular experiments, conjugate 12 can degrade BRD4 in HER2-positive cells BT-474 but cannot degrade BRD4 in HER2-negative cells MDA-MB-231.
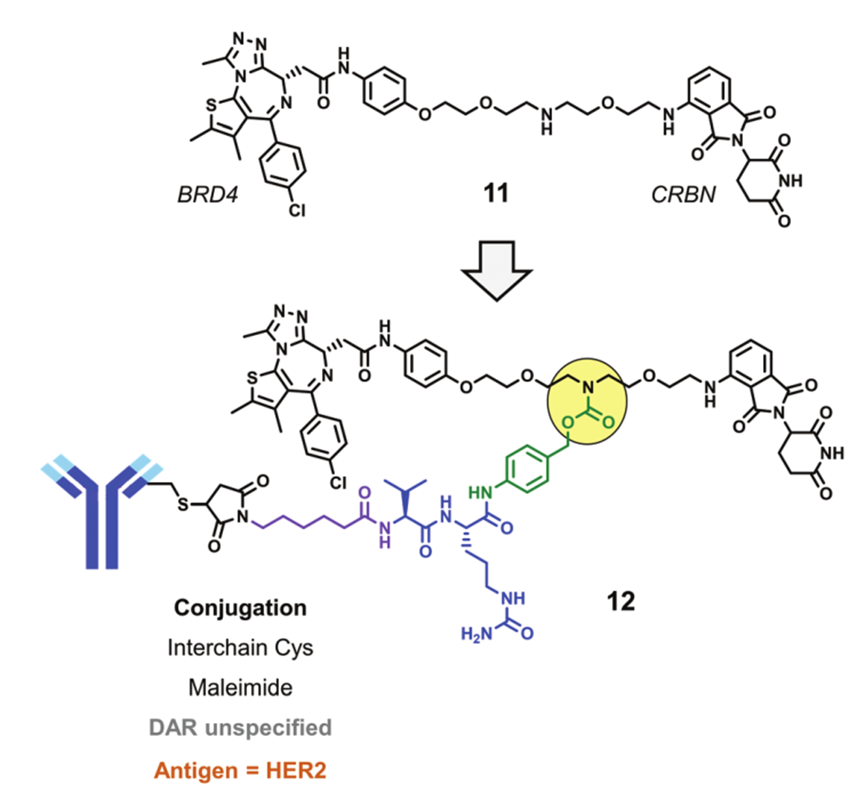
Conjugate 12 (Source: Chemical Society Reviews)
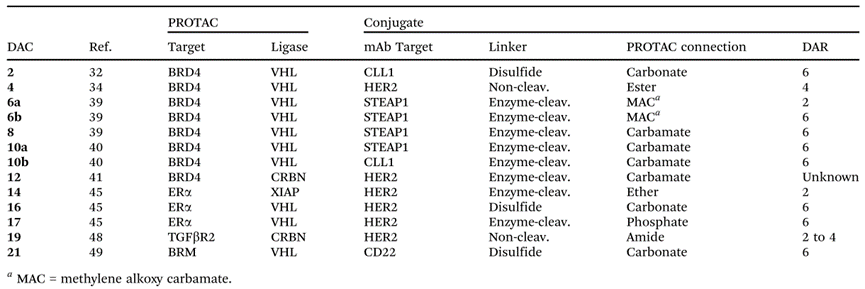
Currently, DACs are still in their infancy, but several candidates with in vitro and/or in vivo biological activity have been identified. The PROTAC molecules in these DAC structures utilize different E3 ligases and target different target proteins. Additionally, various novel linker groups and antibody conjugation technologies have been applied to DACs.
Information on some DAC drugs (Source: Chemical Society Reviews)
The vast majority of DAC molecules have a DAR (6) greater than mainstream ADCs (2-4), but whether this increase is a universal rule for DAC development requires further study. Multiple studies indicate that the activity of DACs depends on cell surface antigens, suggesting that DAC drugs can deliver corresponding PROTACs to specific tumors and/or cells.
Based on the above results, the prospects for the DAC model are bright. However, the following questions still need to be addressed: (1) which PROTAC molecules are suitable for development into DACs; (2) what conjugation methods best preserve (or even enhance) the biological activity of PROTAC molecules.
References:
[1] Peter S. Dragovich. Degrader-antibody conjugates. Chemical Society Reviews. 2022.
[2] Huang Rong et al. Constructing uniform antibodies based on disulfide bond bridging. Nature. 2021.
-
2022 ASCO Abstract Collection (Reply in the background ASCO to obtain a 731-page PDF)
-
100 New Targets(Reply in the background Targets to obtain related Excel)
-
AACR Information Package(Reply in the background AACR to obtain the information collection)
-
Commentary by Zhu Jidong | The Next Frontier in Drug Development: Biomolecular Condensates, Bringing New Opportunities to Undruggable Targets
-
“Originally, there was nothing, where can dust be stirred?” – Also discussing local new drug innovation and anxiety