Using trim-galore to remove low-quality reads and adaptersAdapters: An adapter is a known short nucleotide sequence used to link unknown target sequencing fragments.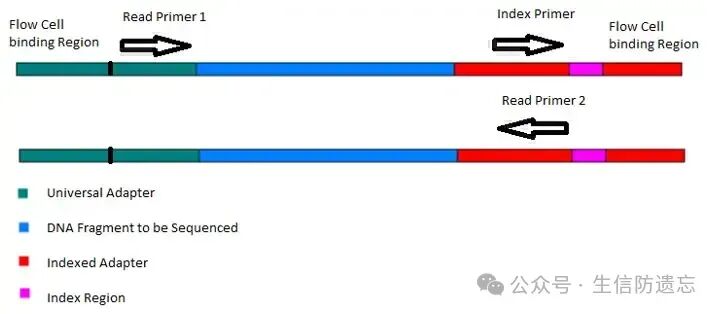 Create a new directory “trim-galore” to store the output results of trim-galore.1. If running a single sample (A-1_1, A-1_2):
Create a new directory “trim-galore” to store the output results of trim-galore.1. If running a single sample (A-1_1, A-1_2):
trim_galore -q 25 --phred33 --stringency 3 --length 36 --paired A-1_1.fq.gz A-1_2.fq.gz --gzip -o ./cleandata/trim_galoredata/-q: Set the Phred quality score threshold, default is 20. For stricter results, change it to 25.–phred33: Generally 33 or 64, indicating the Phred quality score used by the sequencing platform.–stringency: Set the number of overlapping bases that can be tolerated between adapters, default is 1 (very strict). It can be relaxed somewhat, as the second adapter is unlikely to be read by the sequencer.–length: Set the output reads length threshold; reads shorter than this value will be discarded.–paired: For paired-end sequencing results, if one read in a pair is discarded, the other will also be discarded regardless of whether it meets the criteria.2. For multiple samples, write a for loop:Open the file with the vim command to write the for loop
vim trim_galore_batch.sh
for i in A-1 A-2 A-3 A-4
do
trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired /data/RNAseq/${i}_1.fq.gz /data/RNAseq/${i}_2.fq.gz --gzip -o /mnt/d/Users/RNAseq/cleandata/trim_galoredatadone##--paired /mnt/d/Users/RNAseq/${i}_1.fq.gz /mnt/d/Users/RNAseq/${i}_2.fq.gz is the storage location for the results obtained from fastqc, i.e., the qc1 folder##-o /mnt/d/Users/RNAseq/cleandata/trim_galoredata is the output path for trim-galore resultsRun, one file takes about 1 hour
bash trim_galore_batch.shThe resulting files are as follows; the files generated by trim_galore have the suffix _val_1.fq. Then run fastqc again on these fq.gz files (see previous text) to generate .html reports, checking the base quality and adapter removal status after cleaning.Adapters before running trim-galore:
Then run fastqc again on these fq.gz files (see previous text) to generate .html reports, checking the base quality and adapter removal status after cleaning.Adapters before running trim-galore: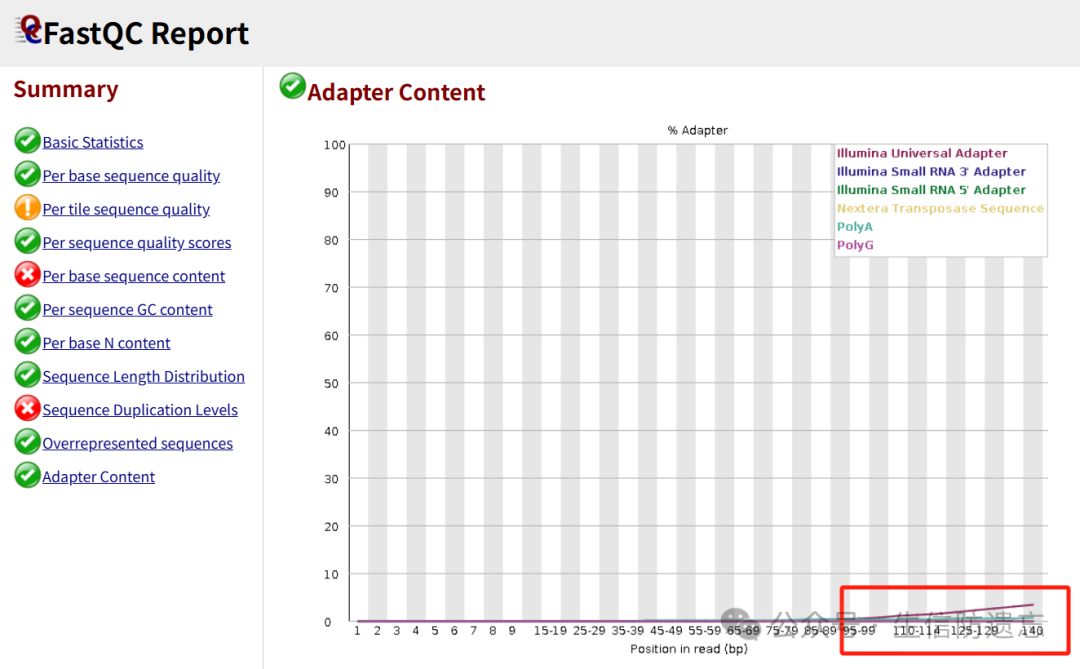 After cleaning:
After cleaning: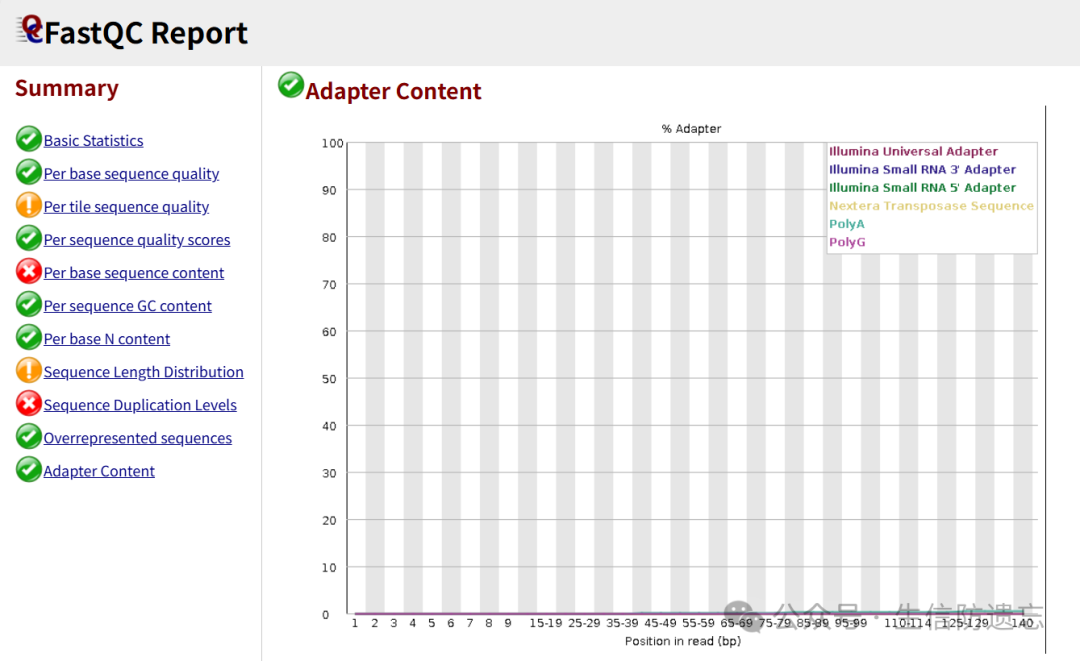 And the base quality is also very high
And the base quality is also very high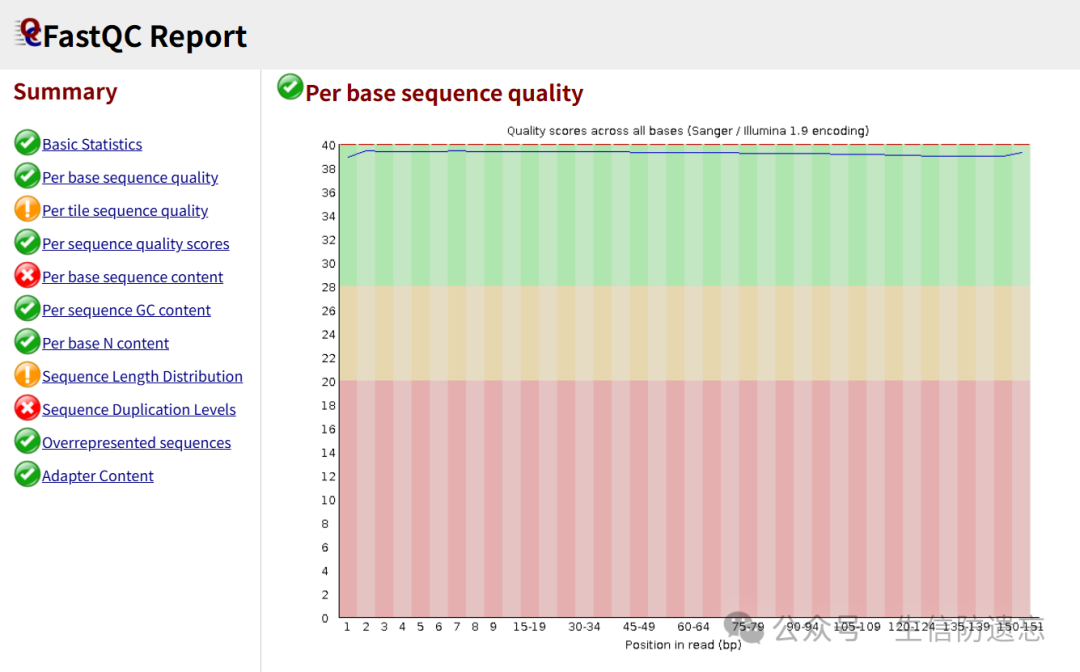 Thus, the data cleaning is complete. Next, use hisat2 to align the sequences to the reference genome.References:1. Jingxuan | What are adapters in high-throughput sequencing? (https://www.jianshu.com/p/3164dca8bd61)2. DoubleHelix | Transcriptome Analysis | Using trim-galore to remove low-quality reads and adapters (https://cloud.tencent.com/developer/article/1703054)
Thus, the data cleaning is complete. Next, use hisat2 to align the sequences to the reference genome.References:1. Jingxuan | What are adapters in high-throughput sequencing? (https://www.jianshu.com/p/3164dca8bd61)2. DoubleHelix | Transcriptome Analysis | Using trim-galore to remove low-quality reads and adapters (https://cloud.tencent.com/developer/article/1703054)