Research Updates
Recently, Professor Cheng Jun and the AI4EC Lab team at Jiajing Innovation Laboratory achieved a significant breakthrough in the field of computational dynamic nuclear magnetic resonance (NMR) spectroscopy of electrolytes using a machine learning combined approach. They successfully predicted the dynamic 7Li NMR chemical shifts in lithium bis(fluorosulfonyl)imide (LiFSI)/dimethyl ether (DME) electrolytes with high accuracy. This achievement represents another iteration of methods and application expansion in the relevant field following the team’s research on dynamic NMR spectroscopy of battery cathode materials and the NMRNet deep learning framework. The related research results were published in the Journal of the American Chemical Society.
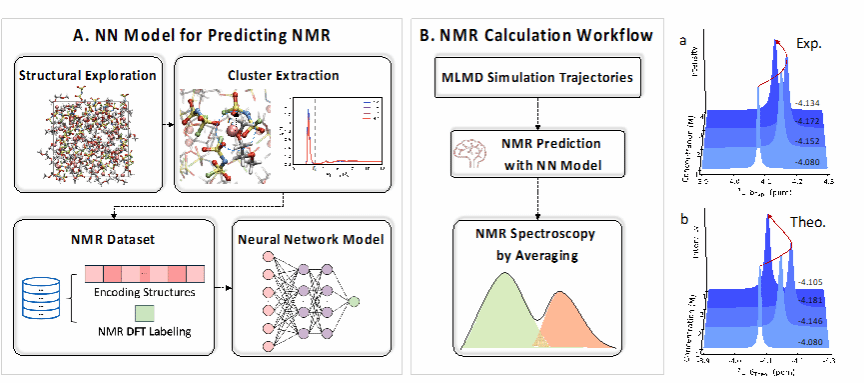
(Left) Method for training the neural network (NN) model. Capturing various structures at different concentrations and extracting the solvation structure of Li+. Subsequently, descriptors are used to encode the structures and calculate their corresponding chemical shifts. (Middle) NMR spectrum prediction process. Molecular dynamics (MLMD) simulations are used to generate trajectories, and then the obtained NMR prediction neural network model is utilized to obtain the NMR spectrum. (Right) Comparison of NMR prediction results with experimental results.
Nuclear magnetic resonance (NMR) is a non-destructive characterization method that is highly sensitive to local structural environments, capable of characterizing specific atomic nuclei’s chemical environments in solvation structures and revealing information such as relaxation times. However, establishing a direct correlation between spectral changes and fine molecular structural variations remains a significant challenge. Although previous studies have explored the relationship between structure and spectra using density functional theory (DFT) static calculations of NMR spectra, the actual measured chemical shifts typically represent a weighted average from multiple local sites, integrating structural distributions and thermodynamic fluctuations, among other dynamic characteristics. This statistical averaging effect significantly complicates signal interpretation and further increases the difficulty of establishing spectral-structural relationships. Molecular dynamics (MD) simulations can capture dynamic structural changes of electrolytes, but in complex systems, configuration sampling is intricate, making it challenging to directly relate molecular structures to experimental spectral observations. While machine learning models have improved the speed of chemical shift predictions, there is currently no clear consensus on the relationship between dynamic structural features of electrolytes and experimental spectral observations, necessitating effective computational methods, and validating the correlation between simulations and experiments is crucial for reliability.
Based on this, the team proposed a machine learning combined approach to predict the dynamic 7Li NMR chemical shifts in LiFSI/DME electrolyte solutions. This method successfully revealed the inversion phenomenon of 7Li NMR chemical shifts, aligning with experimental results. Specifically, as the concentration of LiFSI increased from 1 M to 3 M, the changes in solvation structure caused the NMR chemical shifts to move to higher fields, while at 4 M, they shifted to lower fields. Furthermore, the team quantitatively established the relationship between molecular structures and NMR spectra, providing an in-depth analysis of the attribution of solvation structures. The analysis indicated the presence of two competing local solvation structures, with their dominance alternating as the electrolyte concentration approached its upper limit, leading to changes in 7Li chemical shifts. This work provides a detailed molecular-level understanding of the complex solvation structures detected by NMR spectra, guiding the design of improved electrolytes.
The corresponding authors of this research are Professor Cheng Jun and Associate Professor Tang Fujie, with doctoral student You Qi and postdoctoral researcher Sun Yan from our institute as co-first authors. Associate researcher Wang Feng from the Jiajing Innovation Laboratory provided assistance during the research process. This research was supported by the National Natural Science Foundation (22021001, 22225302, 21991151, 21991150, 92161113), the Central University Basic Research Business Fund (20720220009), the National Key R&D Program (2024YFA1210804), and the Artificial Intelligence Application Electrochemical Laboratory (AI4EC), IKKEM (RD2023100101 and RD2022070501).
[Click to read the original paper at the end]



People from the Department of Chemistry, Xiamen University
Official WeChat Account
Click “View” to let more people from the Department of Chemistry see it
