 ▉ Introduction
▉ Introduction
Antibody-drug conjugates (ADCs) are composed of monoclonal antibodies that specifically target antigens linked to small molecule cytotoxic drugs via linkers, combining the powerful killing effect of traditional small molecule chemotherapy with the tumor-targeting capability of antibody drugs. ADCs consist of three main components: the antibody responsible for selectively recognizing cancer cell surface antigens, the drug payload responsible for killing cancer cells, and the linker that connects the antibody and the payload.
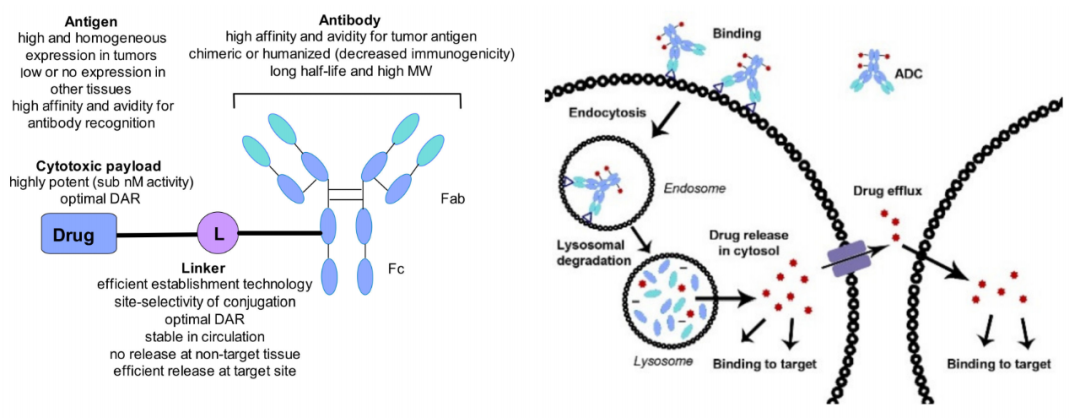
The recognition of antigens by ADCs leads to their internalization into cells via endocytosis. After degradation in lysosomes, the payload is released in a bioactive form to exert its effect, resulting in cancer cell death. The amount of payload inside the cells is determined by the number of surface antigens per cell, the number of drug payload molecules per ADC (also known as drug-to-antibody ratio, DAR), and the time required for antigens to return to the cell surface. The payload may escape after cancer cell death and degradation or may transmembrane out of the cytoplasm. The consequences of this release can be beneficial (also known as bystander effect) or harmful, leading to systemic toxicity.
The first ADC (Mylotarg) was approved in 2000, and since 2019, the number of approved ADCs has more than doubled, with five ADCs approved between 2019 and 2020, indicating a continued hot trend in the field of ADCs.
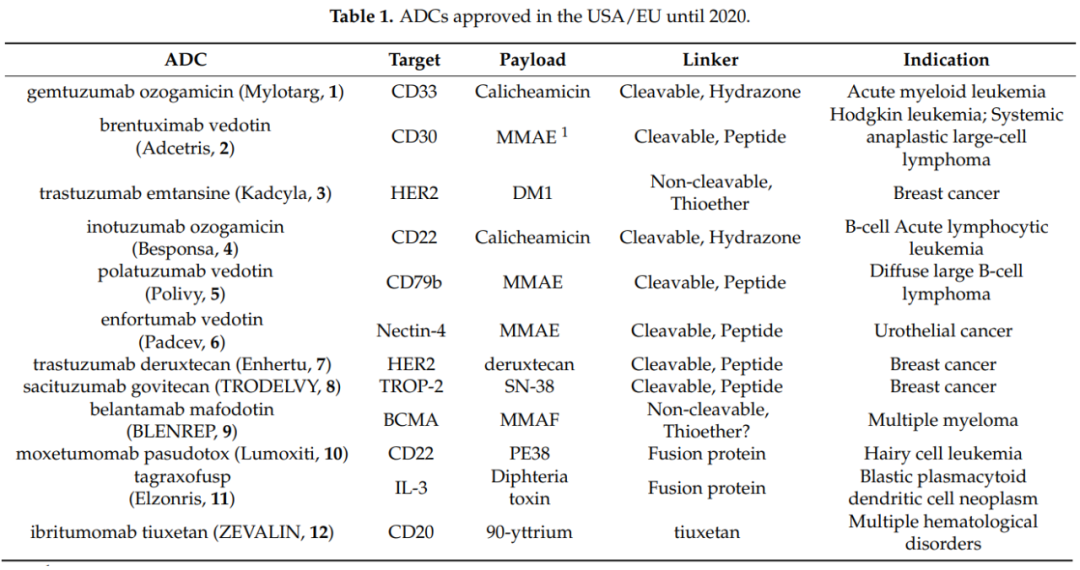
There are several variable components in ADCs, and success clearly does not follow a universal formula. Therefore, how to select the appropriate antibody, the mechanism of antibody endocytosis, where and how to link the linker to the antibody, how many drug molecules to connect to each antibody, how to connect the linker and drug payload, and what the optimal drug payload looks like are questions that require a deep understanding of the biological and chemical properties of the various components of ADCs to obtain satisfactory answers.
1. Microtubule Disruptors
Auristatins
Auristatins are important payloads used in ADCs, with the most famous family member MMAE present in two marketed drugs, Adcetris and Polivy. Currently, more than 10 ADCs with auristatin (such as MMAE) or monomethyl auristatin F (MMAF) as payloads are undergoing clinical trials.
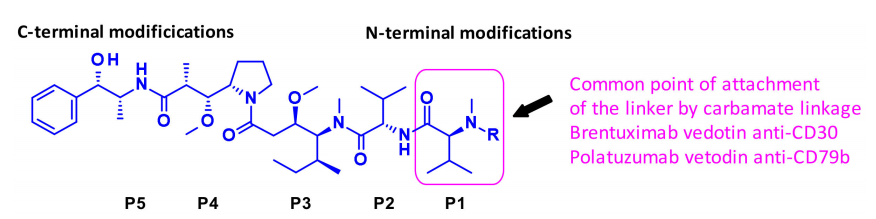
The above figure describes the auristatin and its common linker sites. The structure-activity relationship (SAR) of auristatin has been extensively studied, mainly focusing on the terminal subunits: P1 (N-terminal) and P5 (C-terminal), with the most common approach being the introduction of carbamate functionalities on P1.
In 2015, researchers from Seattle Genetics expanded the range of ADC payloads to include tertiary amines, particularly N,N-dimethyl auristatin, which first linked the drug to monoclonal antibodies through ammonium bonds. The resulting ADCs were stable under physiological conditions, highly active in vitro and in vivo, and exhibited strong immuno-specificity. These results expanded the range of drugs available for targeted delivery using ADCs.
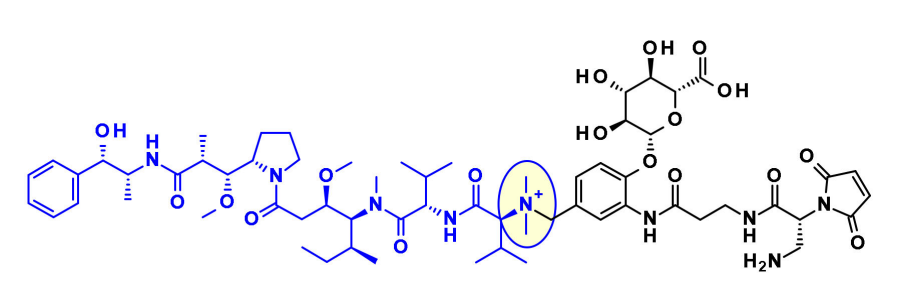
Recently, Agensys introduced azide groups into P2 and P4 subunits by modulating the central subunits P2-P3-P4, producing hydrophilic derivatives with improved potency in vitro and in vivo after coupling with protease-cleavable linkers, providing new avenues for linker connections.
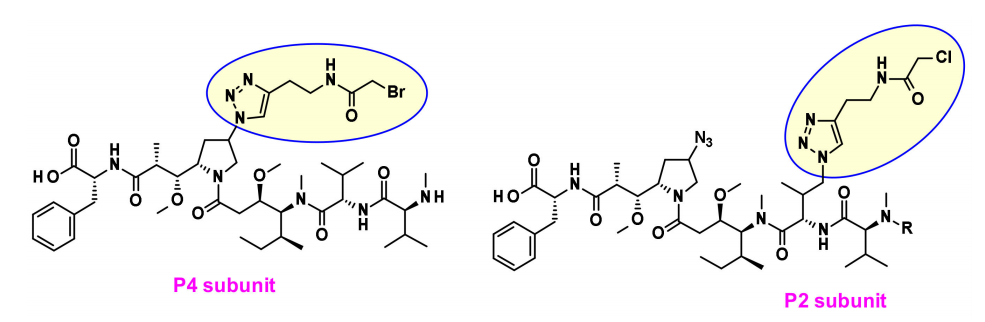
Generally, for auristatins that contain both amines and alcohols, the preferred linker point is the amine coupled via carbamate bonds. Seattle Genetics developed a new strategy to couple alcohol-containing payloads with methylene carbamate (MAC). To stabilize the MAC bond, basic groups and electron-withdrawing groups are positioned close to the amine bond, resulting in a coupling product that is stable under physiological conditions, has high potency, and exhibits immuno-specificity both in vitro and in vivo.
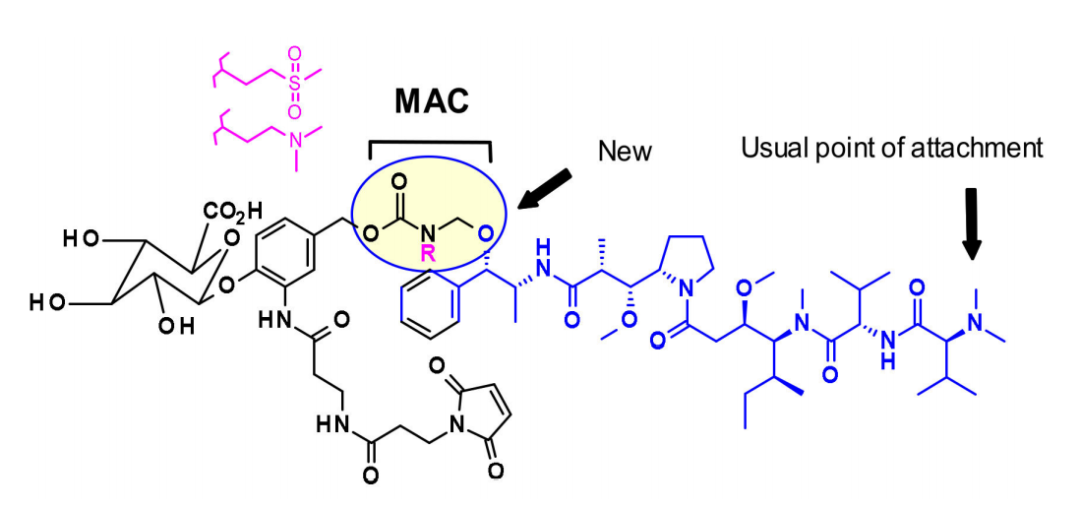
Additionally, researchers at Uppsala University have developed AZASTATIN, a new class of potent auristatin derivatives that include a central amine side chain as an antibody binding site. Their findings confirm that these auristatin derivatives represent a new class of cytotoxic payloads suitable for ADC development.
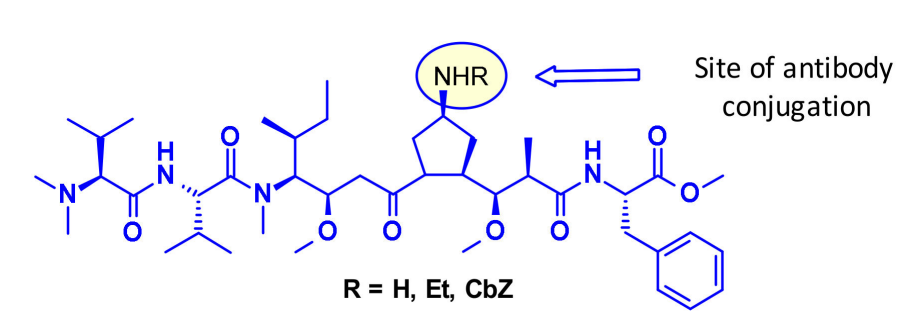
Maytansine Derivatives (DM2, DM4)
Maytansine is a highly effective microtubule assembly inhibitor that can induce mitotic arrest in cells. However, this structure is difficult to conjugate due to the lack of reactive functional groups. To overcome this issue, a series of highly effective derivatives containing SMe groups have been created. The first examples of this class of molecules are DM1 and DM4, which carry methanesulfonyl instead of the natural N-acetyl group.
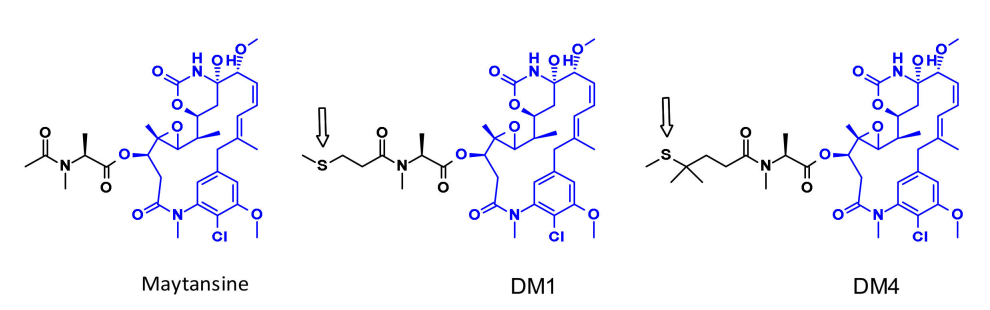
The payloads DM1 and DM4 are coupled with linkers using disulfide bonds. Stable disulfide-linked linkers exhibit good stability in blood circulation while maintaining effective division within the cells.
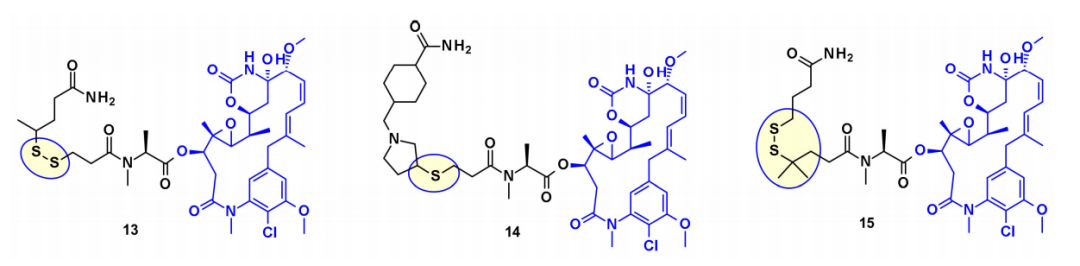
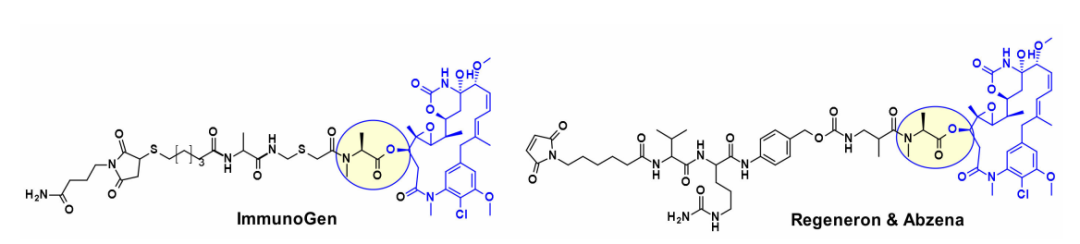
Moreover, several maytansine-based ADCs utilize the same secondary hydroxy group as an attachment point and, in most cases, carry a cleavable linker that is bioconjugated to transglutaminase. For instance, an ADC based on Daratumumab has been shown to specifically deliver DM4 to CD38-overexpressing cancer cells. Recently, ImmunoGen developed a new type of ADC that includes a sulfur-containing maytansinoid linked to the antibody via a highly stable tripeptide linker, with the attachment point being the same as the mentioned hydroxy group. Compared to previous maytansine ADCs, increasing the number of methylene units in the linker increased bystander killing activity and improved efficacy in mouse models. In a similar approach, while maintaining the core macrolide unchanged, researchers from Regeneron and Abzena studied the effects of nitrogen substitution on N-methylalanine and also altered the length of the side chains on the macrolide, as well as the linkers connected by primary and secondary amines.
Microtubule Lysins
Tubulysins are effective inhibitors of microtubule polymerization that can rapidly disassemble the cytoskeleton of dividing cells and induce apoptosis. They are a naturally occurring family of tetrapeptides containing Mep, Ile, Tuv, and Tut, where R3=OH or Tup, R3=H.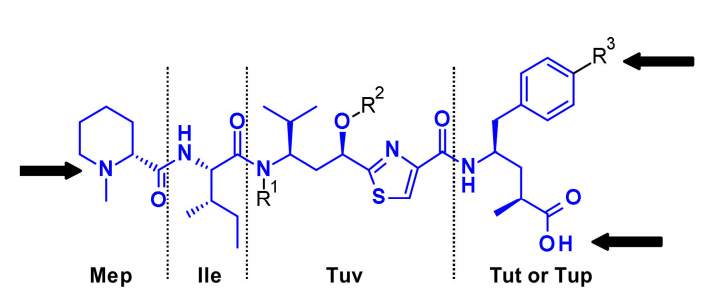
Utilizing tubulysins as ADC payloads, their extensive attachment points have been thoroughly developed. A prominent attachment point in this structure is the carboxylic acid of the Tut or Tup module, as seen in Endocyte’s EC1428, where the carboxylic acid is linked to the linker via an acylhydrazone. Oncomatryx also adopted a similar approach to install a cleavable PABAValCit maleimide linker in the same manner.
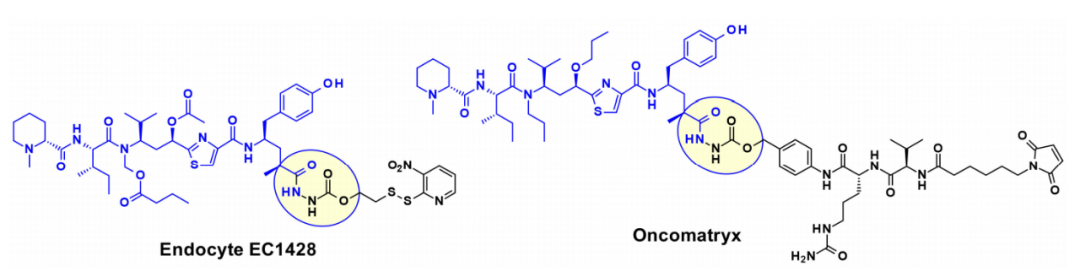
A different method used by AstraZeneca, Bristol-Myers Squibb, and Pfizer relies on the derivatization of the aromatic ring in Tup or Tut.
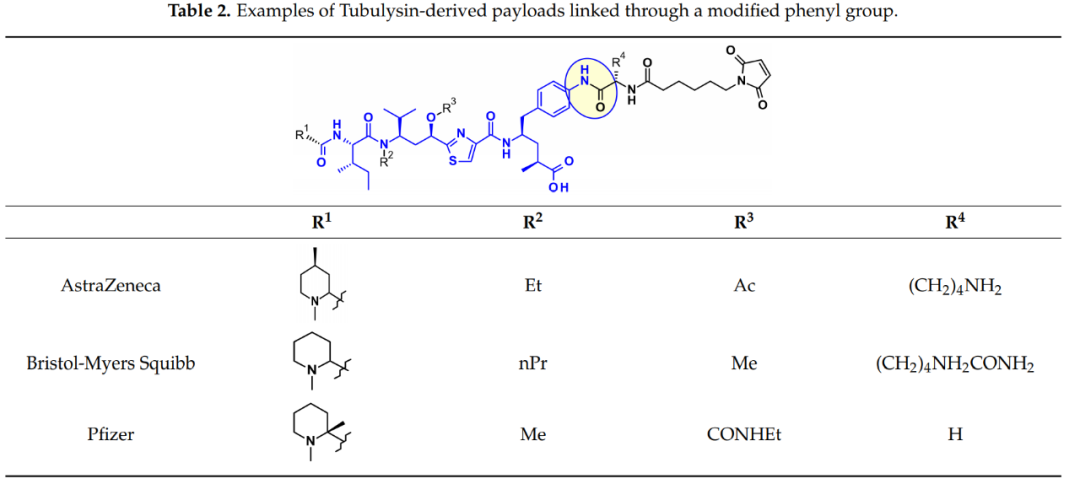
The linkers with the Mep group have also been extensively studied. Ingenica researchers reported that demethylated Mep analogs retained potent cytotoxic activity and could be viewed as valuable payloads, allowing the introduction of non-cleavable maleimide hexyl linkers on secondary amines.
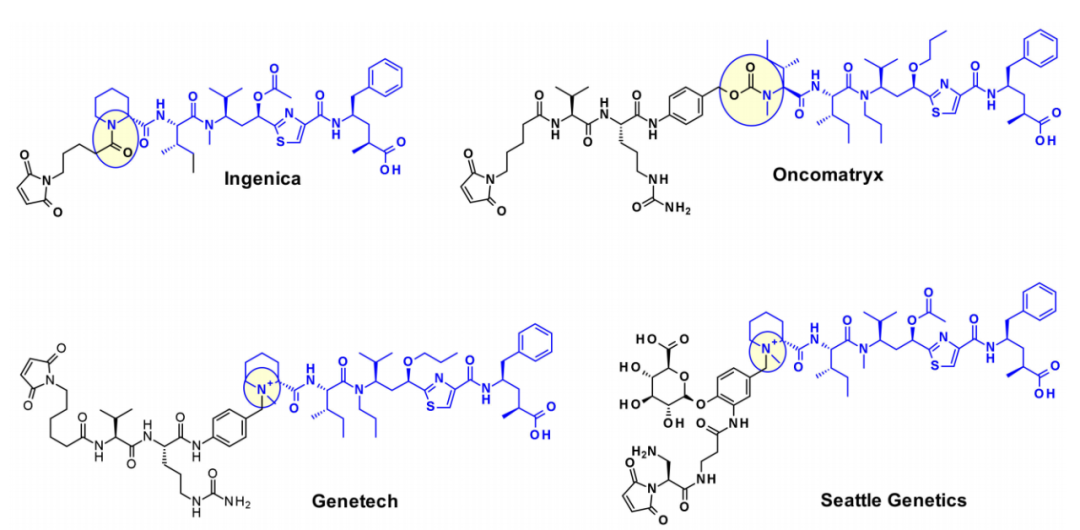
Oncomatryx’s research indicates that when Mep is replaced by another motif with a secondary amine, introducing cleavable linkers through carbamate bonds is an effective route for producing ADCs. Particularly interesting is Genentech’s work on linking linkers to payloads containing tertiary amines through quaternary ammonium groups. Introducing mc-Val-Cit-PABA linkers on the payload leads to more hydrophilic conjugates and improved stability in blood. Seattle Genetics also adopted a similar approach, where overexpressing glucuronic acid improved both hydrophilicity and selective intracellular cleavage through overexpressed β-glucuronidase in cancer cells.
Cryptomycins
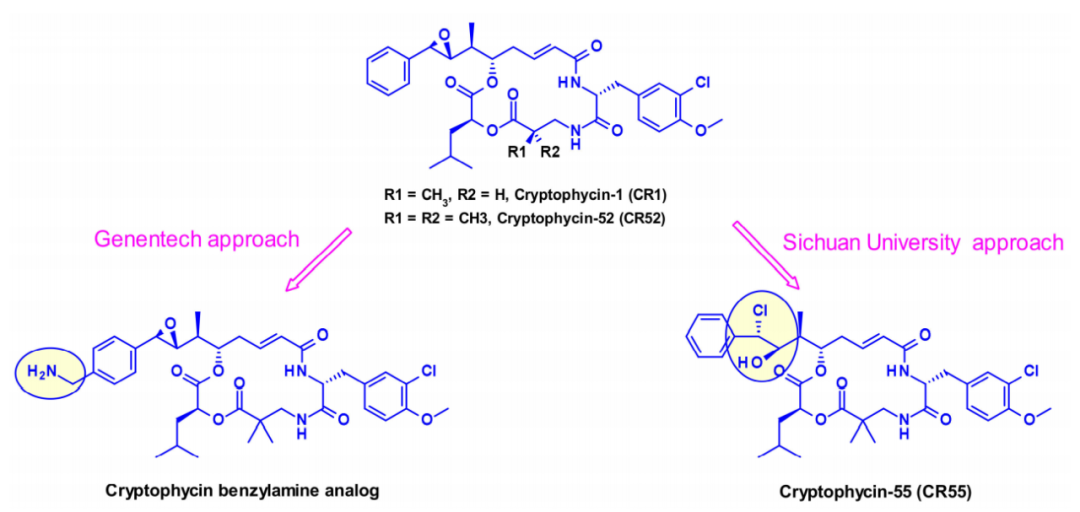
Cryptomycins (CR) are a family of hexacyclic dipeptides with antitumor activity. Results from existing clinical trials indicate that their toxicity levels at doses required to achieve therapeutic effects are unacceptable.
Several groups have attempted to utilize CR for ADCs, but due to the lack of coupling sites in CR, there are currently two different approaches that introduce other groups to enable the coupling of ADC linkers. One is the conversion of phenyl to benzyl by Genentech researchers to produce an effective payload that is suitable for coupling via carbamate bonds. In the second approach, researchers from Sichuan University utilized the prodrug form of cryptomycin-52 (CR55), which can re-cyclize to CR52 under physiological conditions.
Antimicrotubule EG5 Inhibitors
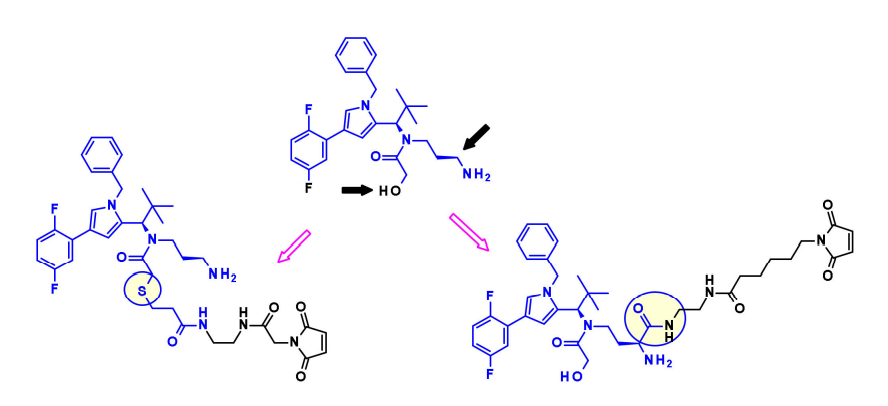
Spindle-driving protein (KSP, also known as Eg5 or KIF11) is an ATP-dependent motor protein involved in the separation of centrosomes during the cell cycle. Therefore, blocking this important event in mitosis with KSP inhibitors (KSPis) can produce antitumor efficacy.
Bayer discovered a new class of KSPis from the pyrrole subclass, investigating the compatibility of the molecule’s different positions with linkers that maintain strong affinity to KSP.
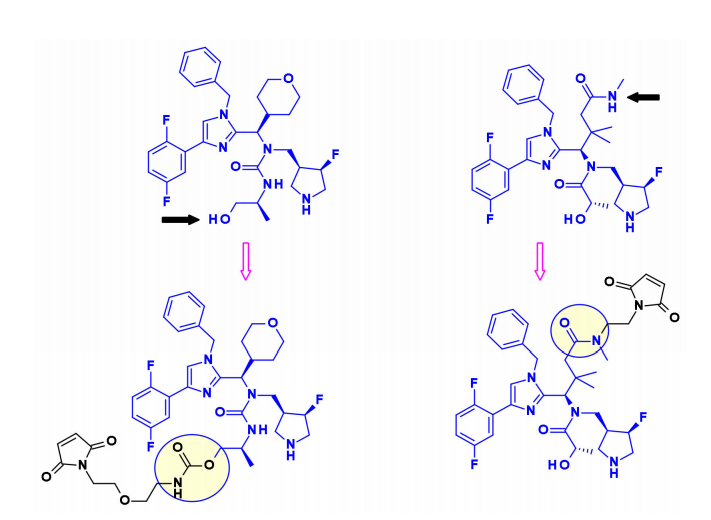
2. DNA Damaging Agents
Pyrrolo-benzodiazepines and Indolyl-chlorobenzodiazepines
Pyrrolo[2,1-c][1,4]benzodiazepines (PBD) are a class of natural products with antitumor activity. Their mode of action involves selective alkylation in the minor groove of DNA, where the N2 of guanine forms a covalent bond with the electrophilic N10/C11 imine on PBD.
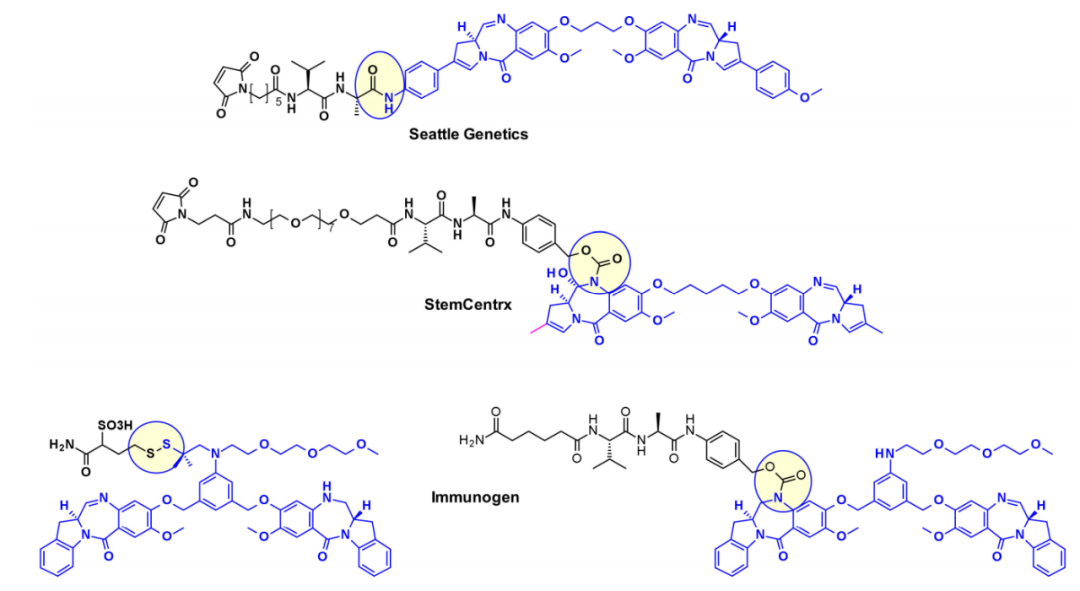
Seattle Genetics utilized the aniline of SGD1882 as an attachment point, mimicking the PAB unit commonly used in cleavable linkers to release free PBD payload. StemCentrx collaborated with Spirogen to connect an amino acid carbamate linker to the N-10 position of PBD. The same carbamate bond was also used by Immunogen for structurally similar indolyl-chlorobenzodiazepine (IBD) payloads. They also reported different methods for the same class of IBD, where a substituted phenyl ring was used as a linker between two IBD monomers at the C8/C8′ positions.
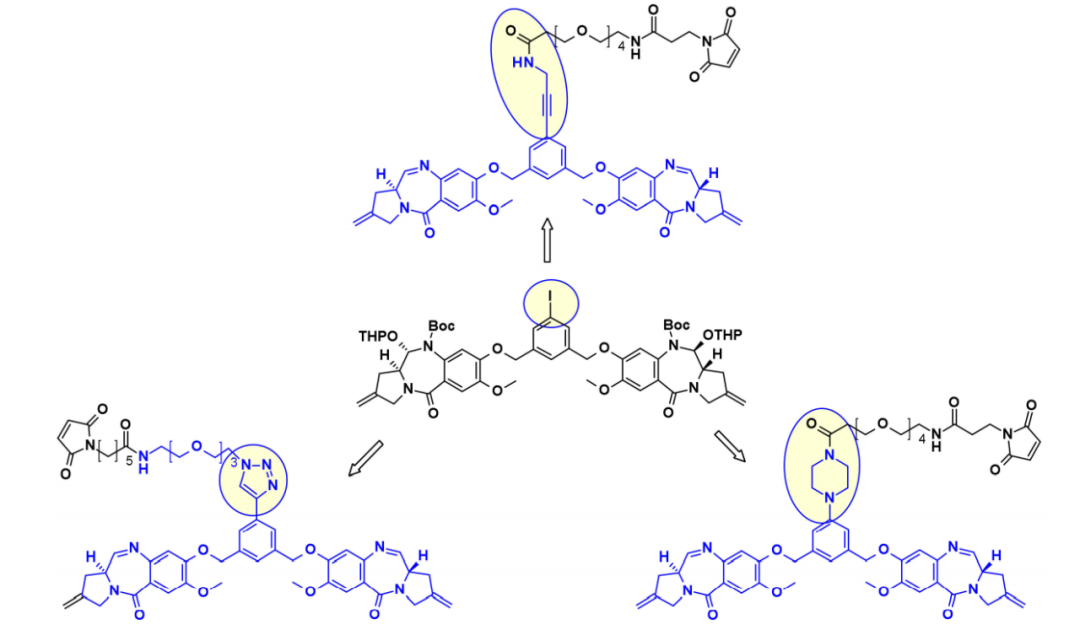
In a similar manner, Spirogen and Genentech designed a PBD with an iodophenyl linker, allowing the introduction of different linkers in transition metal-catalyzed reactions. By employing Sonogashira coupling, Buchwald–Hartwig coupling, or azide-alkyne click reactions, linker-payload conjugates were obtained with alkynes, piperazines, or triazoles, respectively.
Doxorubicin
Doxorubicin is a potent cytotoxic agent that binds to the minor groove of DNA through its highly active cyclopropane ring and alkylates adenine at position N3. The non-cyclized, halomethyl form of doxorubicin shows significantly reduced cytotoxic activity. Due to the phenolic group in the molecule acting as an internal racemization activator, the coupling strategy in doxorubicin ADC development focuses on the linker connecting the phenolic functional group.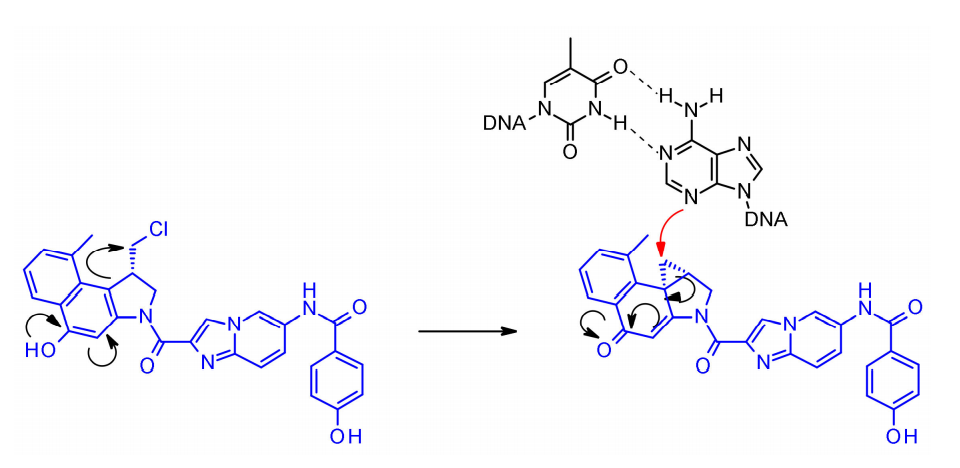
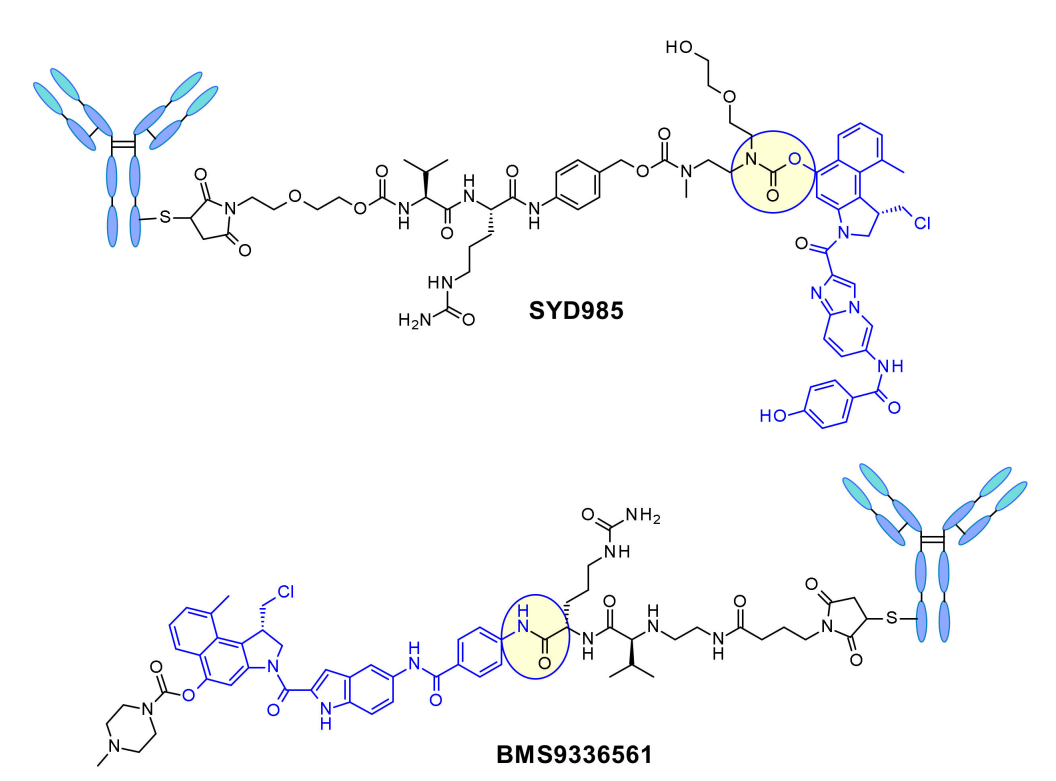
In SYD985 developed by Synthon, the phenolic group is connected to the Mc-val-cit-PABC payload through a diamino carbamate linker. After cleavage by cathepsin B, the free phenol promotes an intramolecular rearrangement to the electrophilic cyclopropyl form. Medarex adopted a different approach by connecting the linker with N-methyl piperazine carbamate portion, masking the phenolic prodrug. In vivo, the phenol will be released, followed by the active cyclopropyl being formed under the action of carboxylate esterase.
Camptothecin
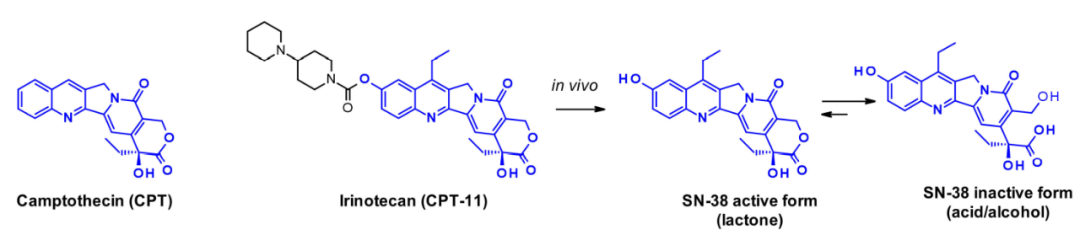
Camptothecin (CPT) and its derivatives are classic examples of topoisomerase I inhibitors. They stabilize the DNA single-strand breaks induced by topoisomerase, leading to double-strand breaks when the ternary DNA-TOP1-inhibitor complex encounters a replication fork. Natural camptothecin is a pentacyclic structure whose extremely low solubility prevents its widespread use as an anticancer drug. Its water-soluble prodrug irinotecan has been licensed for metastatic colorectal cancer. SN-38 is an active metabolite of irinotecan, generated in the human liver by carboxylate esterase, which can be inactivated by opening the lactone ring.
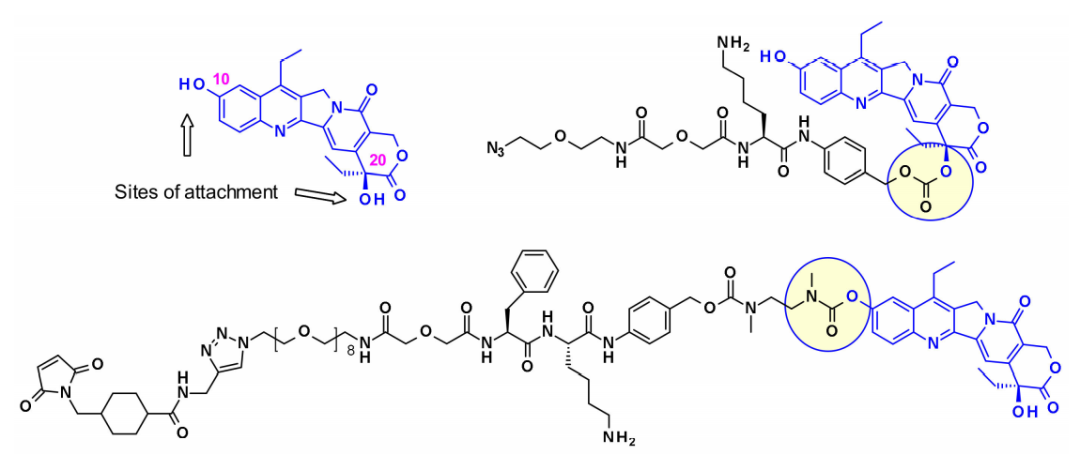
Immunomedics established two different strategies for linking SN-38 through its hydroxyl portion. In one example, the linker is connected through a more reactive C-10 phenolic group, producing a stable carbamate bond, while in another example, the C-20 hydroxyl is used, maintaining the lactone form while being crucial for in vivo efficacy.
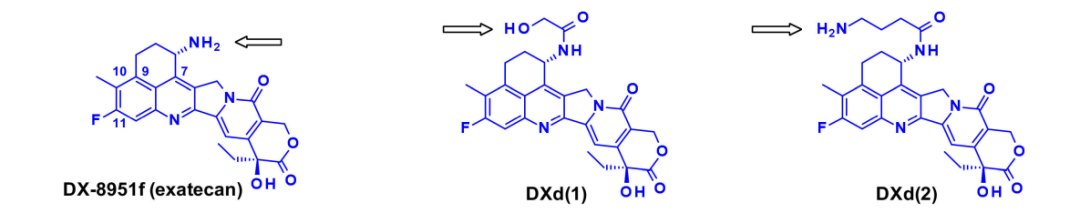
Another highly effective drug suitable for ADCs is exatecan (DDX-8951f). This camptothecin analog features an amine substituent on its cyclohexane ring bridging positions 7 and 9. The amine of exatecan contributes to its water solubility, while the rigidity imparted by the cyclohexane ring is believed to favor the balance between the active lactone form and the inactive hydrolyzed hydroxyl acid. The amine hydroxyl acylation generates DXd (1), while 4-amino-butyric acid acylation generates DXd (2), both retaining the biological activity of exatecan.
Suspended hydroxyl and amine groups are obvious attachment points that can be linked to payloads using enzyme-cleavable Gly-Gly-Phe-Gly peptide linkers. The ADCs generated by coupling to anti-HER2 antibodies show great potential in clinical settings against HER2-expressing cancers.
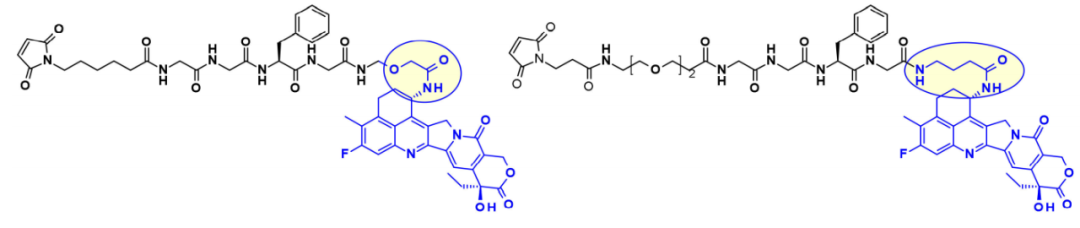
While the cyclohexyl ring of DXd is believed to stabilize the biologically active lactone form, it carries a chiral center, complicating synthetic work and SAR studies. To overcome this difficulty, researchers at Immunogen studied a new set of camptothecin analogs that can couple with monoclonal antibodies. Here, the ring is opened, eliminating the additional chiral center. Starting from a common intermediate, researchers attempted three types of structures and subsequently used different polyphenol linkers to attach the payload. When coupled with antibodies against human epidermal growth factor receptor (HuEGFR), the resulting ADC demonstrated efficacy in the EGFR-positive HSC-2 tumor xenograft model.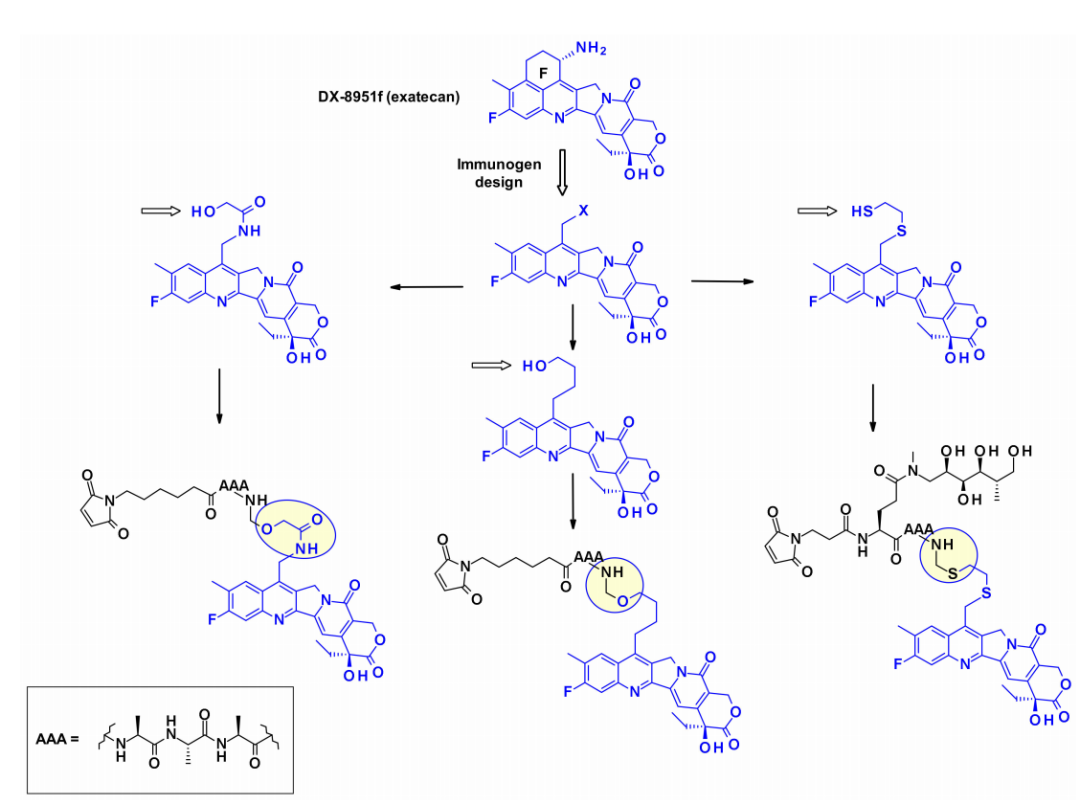
Calicheamicin
Calicheamicin is a class of extensively studied enediyne antibiotics, whose structure and mechanism of action are particularly interesting and complex, making them a class of antibiotics in the field of ADC payloads. The strategy for linking calicheamicin in ADCs is exemplified by the marketed ADCs Mylotarg and Besponsa.
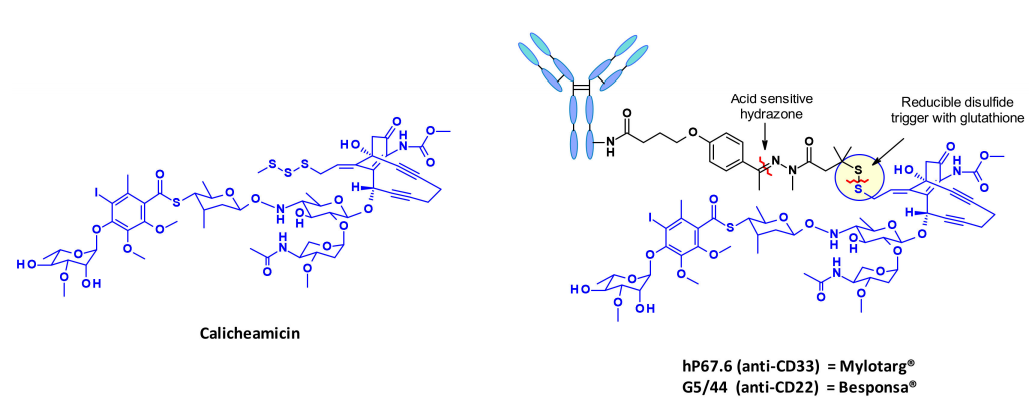
The release of the payload occurs in two steps: sensitive cleavage of the hydrazone in the acidic intracellular environment, followed by the reduction of the disulfide bond by intracellular glutathione. The released thiol undergoes intramolecular 1,4-addition of the enediyne, triggering a Bergman cyclization reaction that generates a diradical. This active intermediate can abstract hydrogen atoms from the deoxyribose backbone, resulting in double-stranded DNA breaks and consequently leading to cell death.
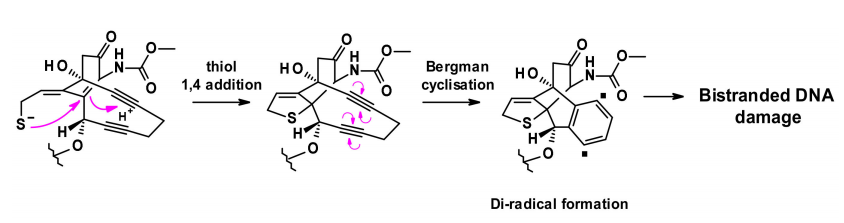
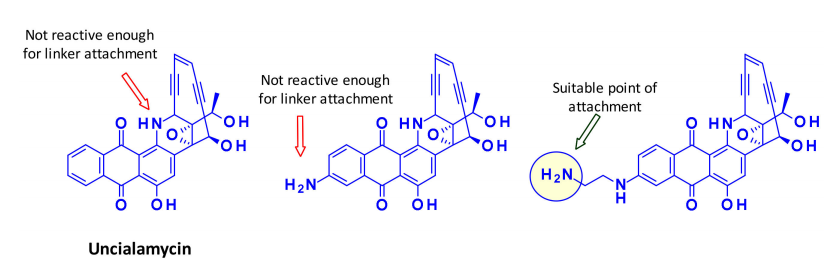
Recently, a new enediyne natural product called uncialamycin was isolated from a strain of streptomyces discovered in British Columbia. This structure has been confirmed through total synthesis, and since then, several highly efficient synthetic analogs have been prepared as potential effective payloads for ADCs.
BMS researchers indicated that due to low reactivity under various peptide coupling conditions, the secondary amine of uncialamycin is not a suitable attachment point. They synthesized an analog where one amine is directly introduced onto the aromatic ring, but this phenylamine’s reactivity is also too weak to serve as a group for linker introduction. On the other hand, using an aminoethyl extension provides a suitable attachment point for the linker.
From the latter effective payload, they prepared precursors using protease-cleavable dipeptides and non-cleavable linkers. CD70-ADC has a cleavable linker and exhibits highly specific cytotoxic activity against renal cancer cell lines, while the corresponding non-cleavable ADC shows no activity in the same cell line.
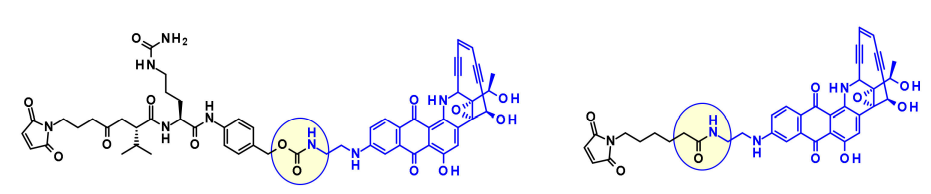
Recently, to continue this work, BMS researchers used phenolic groups as attachment points in designed, efficient, and chemically stable uncialamycin analogs. Using newly developed phenolic alkylation, a classic cleavable linker was added to the phenolic group of the payload. The resulting payload is conjugated to antibodies, showing antigen-specific antitumor activity both in vitro and in vivo.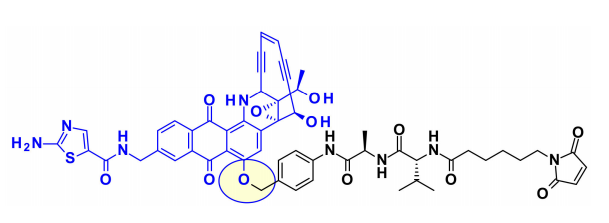
3. Innovative Drugs
Apoptosis Inducers (Bcl-xL Inhibitors)
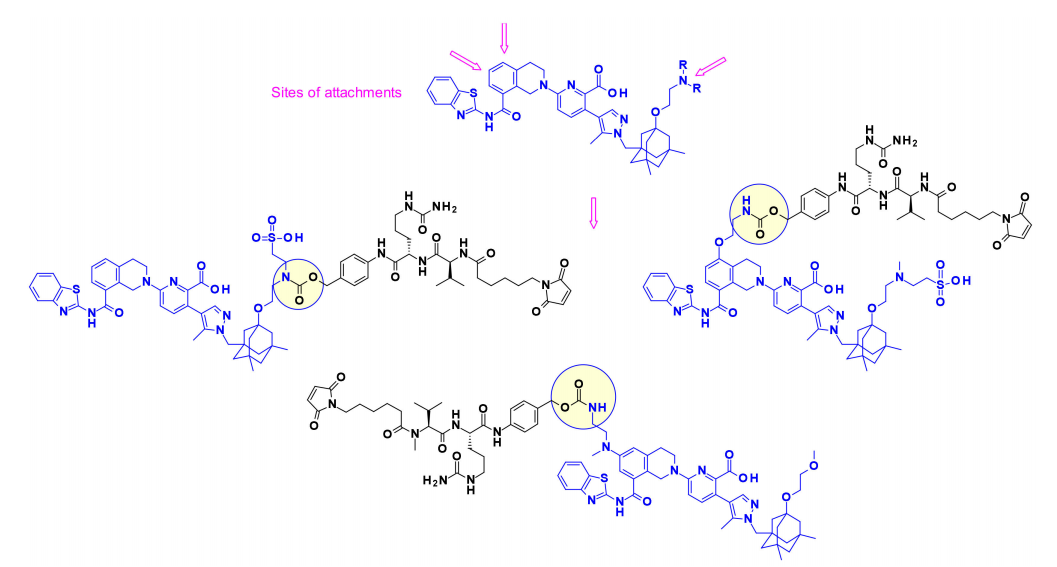
Overexpression of anti-apoptotic Bcl-2 family members (including Bcl-xL) is one of the mechanisms by which cancer cells acquire resistance to apoptosis. Drugs that can block the BH3 binding domain on Bcl-xL can trigger apoptosis in cancer cells. In 2017, AbbVie first demonstrated the payload of BcL-xL inhibitors in the form of ADCs, targeting specific cells or tissues that express EGFR. Interestingly, researchers used three different attachment points on the payload to connect cleavable linkers. Aminoalkyl extensions of the core modifications were used to establish suitable attachment points when needed.
Thailanstatin and Its Analogues
Targeting spliceosomes is a promising therapeutic option for targeted cancer therapy. Several natural products can inhibit RNA splicing by binding to different spliceosome subunits. The most representative is thailanstatin A, which binds to the SF3b subunit of the spliceosome, thereby blocking RNA splicing.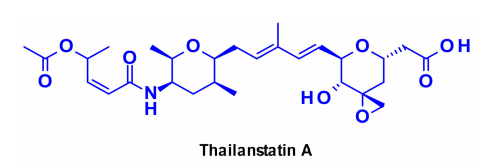
Thailanstatin A lacks a group suitable for linker attachment. To address this issue, a carboxylic acid was coupled with ethylenediamine to introduce an amine-containing spacer, which is commonly used for linker installation.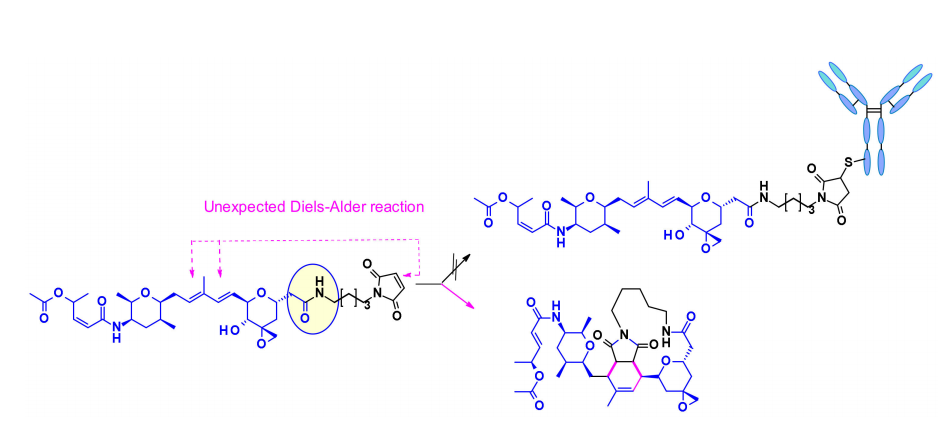
Another difficulty in using this natural product for ADCs is the presence of multiple reactive functionalities. For example, the diene in the central core can react with the maleimide portion used for bioconjugation through Diels–Alder reactions. This issue was resolved by using another conjugated portion, halogenated acetamides. Combining these two modifications and including cleavable linkers has been reported for the first time in patent literature, claiming that they exhibit moderate activity in several HER2-expressing cell lines.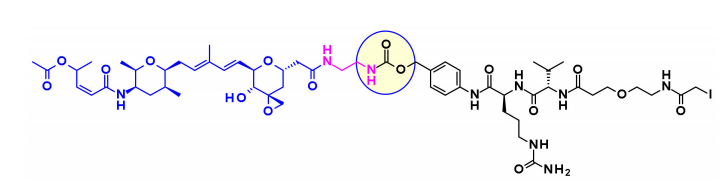
Recently, Pfizer reported that direct coupling of the carboxylic acid with the effective surface lysine of the antibody (without linker coupling) resulted in the most effective Thailanstatin ADC to date. The activity of these lysine conjugates is related to the drug load, while this characteristic has not been observed in other classes of payloads. ADCs show good efficacy in gastric cancer xenograft models.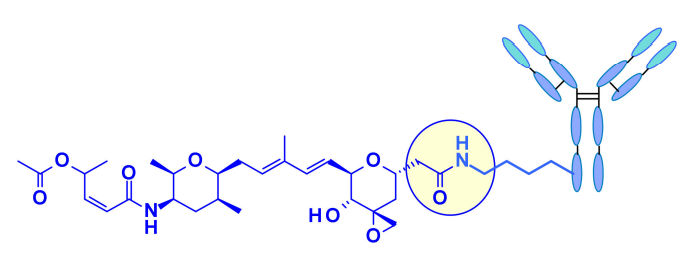
Amatoxins
In the field of ADC technology, the use of transcription inhibitors similar to amatoxins is a relatively new approach. Nine naturally occurring amatoxin derivatives share the same skeletal structure, a large ring composed of eight L-configured amino acids, connected by sulfone portions between tryptophan and cysteine residues. Three side chains of amatoxins are hydroxylated, and the OH groups exhibit good water solubility and bind to target molecules. Two peptides, α-amanitin and β-amanitin, account for 90% of all toxins.
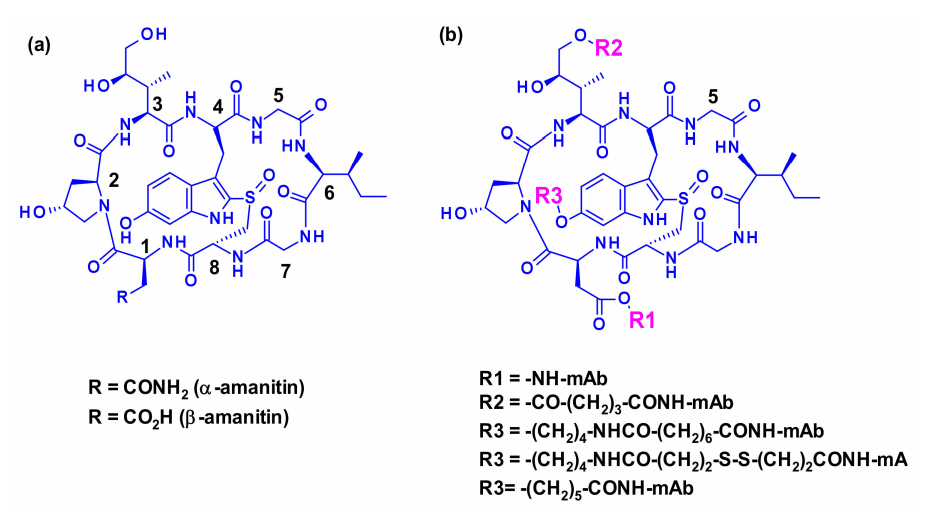
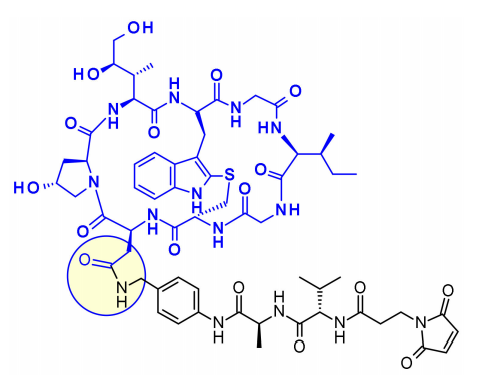
Three attachment points have been used on amatoxins to produce ADCs. The first attempt was to couple the carboxyl group of β-amanitin to the amino group of lysine on IgG, which had good plasma stability and high cytotoxicity, but the yield of this bioconjugate was low. The hydroxyl of dihydroisoleucine was also considered as an attachment point, introducing glutathione as a linker, followed by coupling through lysine, resulting in ADCs with excellent in vitro cytotoxicity and in vivo antitumor activity, but unfortunately, the serum carboxylesterase cleavage of the linker resulted in poor circulation stability. The third method, attachment to the 6-hydroxyl of tryptophan, represents the current standard procedure, where phenol is etherified with various linkers.
Based on the findings, a new class of ADCs has been developed that utilizes a specific linker for the effective payloads of amatoxins, which include a phenolic group as an attachment point. The resulting payloads exhibit excellent stability and potency both in vitro and in vivo, demonstrating the potential of amatoxins as effective payloads for ADCs.
Recent Advances in ADC Clinical Development
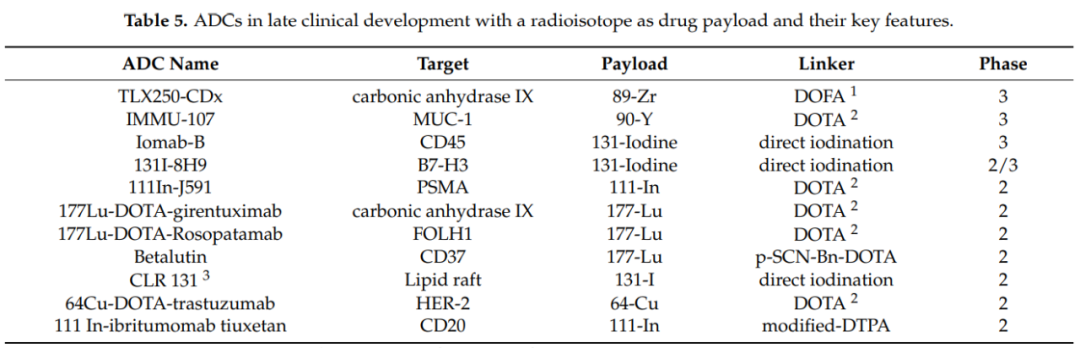
Currently, the field of ADC drug development is thriving, with rapid advancements in effective payloads, linkers, and conjugation technologies leading to higher uniformity, stability, and production efficiency of ADCs. This fuels the clinical development of ADC drugs. The following three tables summarize ADCs in late clinical development as of February 2021, categorized by “small molecules, biomacromolecules, and radioactive isotopes.”
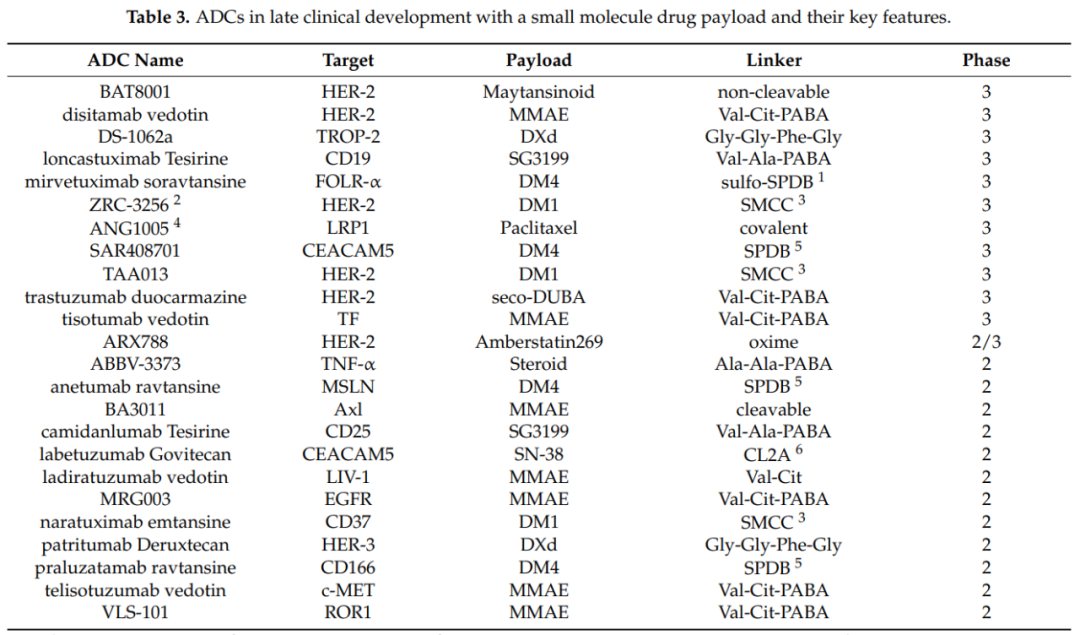
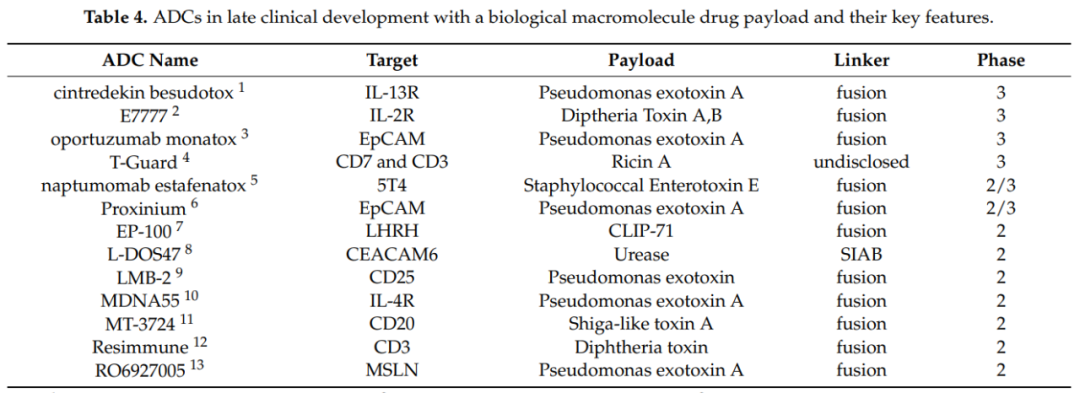
Although the first ADC was approved by the FDA over 20 years ago, the pharmaceutical industry has had to undergo a long and tedious learning process to establish a stable ADC pipeline in the market and clinical development. Despite the current 11 approved ADCs, the field is experiencing an explosion due to technological advancements in ADCs, with multiple breakthroughs in payloads, linkers, and conjugation technologies, further deepening the comprehensive understanding of this new class of drugs.
Furthermore, determining the drug dose-exposure-effect relationship is key to the success of ADCs, with endocytosis being a critical part of this relationship, essential for optimizing dosing regimens to maximize therapeutic indices. However, despite the current fervor in the ADC field, we know little about the endocytosis of target receptors. Additionally, many core components and key effectors of endocytosis are crucial, but these proteins may be universally mutated in cancer, which may also affect ADC endocytosis and efficacy. It is believed that as the field develops, with countless experiences accumulated, ADCs will eventually usher in a true spring.
References:
1.The Chemistry Behind ADCs. Pharmaceuticals (Basel). 2021 May; 14(5): 442.
 Scan the WeChat QR code to add the editor of the bioproducts circle, and those who meet the conditions can join the bioproducts WeChat group!
Scan the WeChat QR code to add the editor of the bioproducts circle, and those who meet the conditions can join the bioproducts WeChat group!
Please indicate: Name + Research Direction!


All articles reproduced by this public account are for the purpose of conveying more information, and the sources and authors are clearly indicated. If any media or individual does not want to be reproduced, please contact us ([email protected]), and we will delete it immediately. All articles only represent the author’s views and do not represent the position of this site.
