 –01–
–01–
Introduction
For decades, advancements in antibody-drug conjugates (ADC) and bioconjugates have revolutionized targeted therapies for various diseases. Unlike traditional chemotherapy, ADCs selectively target tumor cells while sparing normal cells, making them highly suitable for cancer treatment. Currently, there are 17 types of ADCs approved by regulatory agencies in different countries worldwide. Over 200 more ADCs are being explored in clinical trials, with expectations for more ADCs to be commercialized in the coming years, benefiting cancer patients.
Several factors in the design of ADCs influence their success, including antibody, linker, payload selection, drug attachment sites, and drug-to-antibody ratio (DAR), all of which play crucial roles. The method of drug attachment to the antibody is critical; the location and manner of attachment can affect the stability, efficacy, and pharmacokinetics (PK) of the ADC.
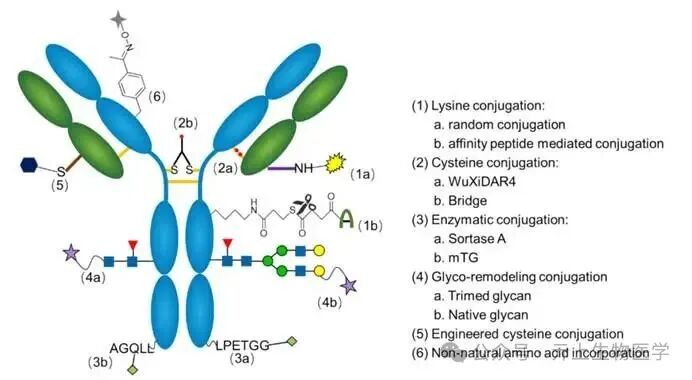
Conjugation methods are traditionally divided into two categories: random conjugation and site-specific conjugation. Further, these techniques can be classified into three categories. The first category is random conjugation (e.g., random lysine). The second category is site-specific but not site-selective, as these sites are limited by the conjugation technology used. This category includes enzyme, glycan, affinity peptide conjugation, and “random” interchain cysteine conjugation. The third category is site-specific and site-selective, modifying amino acid sequences, such as engineered cysteine or non-natural amino acids, providing various binding sites. Regarding CMC, random conjugation and interchain cysteine conjugation are generally simpler. Site-specific and site-selective conjugation techniques offer better homogeneity but may encounter more CMC challenges, including protein expression issues (e.g., non-natural amino acids), additional purification steps required to remove impurities (e.g., enzymes), and extra analytical and quality control challenges (e.g., DAR distribution).
–02–
1. Random Conjugation
Lysine conjugation is a mature technology for producing ADCs, utilizing approximately 40 solvent-accessible NH2 groups on lysine residues, which are highly nucleophilic in neutral solutions. Electrophilic reagents primarily target these amines, allowing linker payload attachment without altering the antibody itself. To date, five commercially available ADCs have demonstrated the efficacy of lysine conjugation. Encouraged by this success, a range of broader linkers has been developed, including N-hydroxysuccinimide (NHS), its analogs, benzoyl fluoride, isothiocyanates, and maleimide esters.
However, due to the high reactivity of these linkers with heteroatoms, they can also be quenched in water. In fact, the hydrolysis of NHS esters is one of the key factors affecting the linker-payload (LP) equivalence in lysine conjugation. Furthermore, less reactive linkers can react with cysteine’s -SH residues under mild conditions, where the reaction with lysine is slow.
Overall, lysine conjugation remains a reliable method for producing ADCs, yielding chemically stable and reproducible products. Additionally, the development of site-specific binding techniques continues to enhance its potential for better homogeneity.
–03–
2. Site-Specific but Non-Selective Conjugation
Interchain cysteine conjugation
Typically, IgG1 antibodies contain four pairs of interchain disulfide bonds in solvent-exposed regions. After reduction using reducing agents such as tris(2-carboxyethyl)phosphine (TCEP) and DL-dithiothreitol (DTT), eight free thiols can be obtained. Compared to reactions on lysine residues, thiols are softer nucleophiles and more prone to undergo Michael addition reactions. This property allows maleimides and their analogs to be used as linkers, enabling the formation of clean and nearly quantitative thioether bonds. This bioorthogonal chemistry is well-suited for antibody modifications, producing variants with 0, 2, 4, 6, and 8 payloads.
Due to fewer thiols, cysteine-mediated ADC heterogeneity is much lower than that of lysine methods. Compared to other conjugation methods, maleimide conjugates stand out for their simplicity, controllable conditions, and high yields. To date, maleimide conjugation with reduced interchain disulfides remains a primary method for constructing ADCs. Among 15 commercialized ADCs with potent toxic payloads, 10 and most of the ADCs in clinical stages have utilized maleimide conjugation technology.
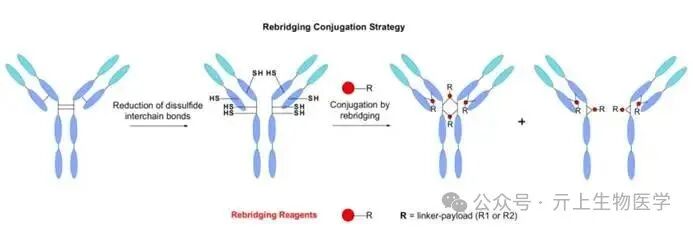
Maleimide-thiol conjugates are prone to reverse Michael addition reactions, leading to premature release of the payload through serum protein interactions. This reverse Michael addition reaction can affect the stability of ADCs in plasma, reducing their efficacy and safety. To address this issue, scientists have explored several strategies, including catalyzing the hydrolysis of maleimides by introducing additional groups such as N-aryl and ortho-amino, or replacing maleimides with open-ring maleimide esters to avoid reverse Michael addition reactions. Additionally, novel linkers such as KTHIOL™, P5™, and bromoacetylamino decanoic acid have shown enhanced thiol selectivity and resistance to reverse Michael addition reactions.
Enzyme Labeling Conjugation
Enzyme conjugation involves directly attaching the payload to the antibody by utilizing enzymes that recognize specific amino acid sequences. This technology provides high uniformity in ADCs, demonstrating its potential as an effective conjugation method. Many enzymes require sequence engineering and structural adaptability of substrates like antibodies. Enzymes used clinically include Sortase A (SrtA), a 30 kDa transpeptidase that can cleave the LPXTG sequence to form thioester acyl-enzyme intermediates, allowing the peptide LPXT to transfer to the substrate’s N terminus. Notable examples include NBE-002 (SMAC Technology™), currently in Phase 1/2 trial stages.
Another is formylglycine-generating enzyme, which attaches to the CXPXR sequence, converting cysteine to formylglycine. ADCs using this conjugation method include TRPH-222 (SMARtag™), currently in Phase 1 clinical stages. Farnesyltransferase modifies antibodies by adding farnesyl groups to cysteine residues within the CaaX tag, with ADCs like FS-1502 (ConjuAll™) currently in Phase III clinical stages. Other enzymes, such as microbial transglutaminase (mTG), target natural sites on antibodies, such as the Q295 site, catalyzing the formation of amide bonds between the γ-carboxamide of glutamine and the free amine of the payload, with representative molecules like DP303, currently in Phase III clinical stages. Additionally, some ADCs in preclinical research utilize enzymes such as peptide asparaginyl ligase, tubulin tyrosine ligase, trypsin, phosphopantetheine transferase, SpyLigase, and O6 alkylguanine DNA alkyltransferase.
The enzyme conjugation process is generally more complex than traditional techniques, requiring a broader range of materials and more intricate steps, which can significantly impact production costs and the likelihood of success. Furthermore, enzyme conjugation introduces additional components, such as binding enzymes, cofactors, and expression-related impurities, all of which have potential immunogenicity. Additional measures are needed to eliminate catalytic enzymes and contaminants from the final product. Overall, enzyme conjugation is a powerful site-specific technology that has achieved many successes in preclinical and clinical stages. Next-generation enzyme conjugation technologies can improve product uniformity through technological updates and optimized CMC processes.
Glycan Remodeling Conjugation
In the past decade, glycan conjugation has attracted significant interest from academia and industry. While most ethylene glycol conjugation methods still rely on enzymes to facilitate the conjugation process, they do not require amino acid sequence engineering. Early techniques involved using sodium periodate (NaIO4) to oxidize cis-diol groups on glycans, creating aldehyde groups for subsequent modifications. Recent strategies emphasize glycan remodeling, where natural glycans (attached at the N297 site) are modified or replaced with new glycans, which can then be linked to functional linkers or linker-payload complexes.
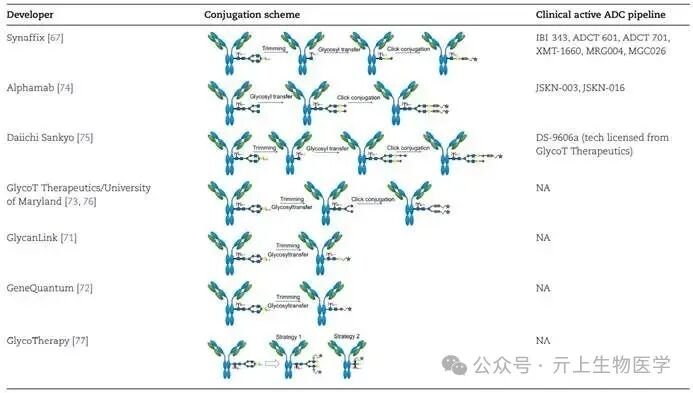
Synaffix’s GlycoConnect™ technology uses endoglycosidases to trim natural glycan isomers, followed by the addition of azide-modified galactose residues and galactosyltransferase. This modified glycan is then conjugated to the payload via click chemistry with compatible handles. This method has been widely applied in ADC development, with six active clinical stage projects utilizing this approach, including IBI-343. However, this process involves at least two enzymes and three conjugation steps, which may complicate development, reduce yields, and increase CMC costs.
Additionally, endoglycosidases found in Streptococcus pyogenes, such as EndoS and EndoS2, can hydrolyze the N-glycans of IgG, making the hydrolyzed residues effective sites for bioconjugation, allowing target molecules to attach to the N297 site in a single conjugation step. This method helps to homogenize the glycan structure of monoclonal antibodies, and it is also applicable to any IgG subtype. Although not yet clinically validated, the endo-S2 method is highly attractive due to its reduced CMC costs.
Affinity Peptide Conjugation
Affinity-directed conjugation utilizes affinity peptides from protein A or G IgG binding sites. These peptides selectively bind to specific sites on the Fc region, bringing them closer to certain lysine residues and enhancing the reaction rate between the linker and -NH2 groups. To prevent side reactions with other reactive -NH2 groups, mild linkers should be used. Typical binding sites are located near the K248/K288 or K337 residues of the Fc region; while sequence modifications of the affinity peptide can shift the binding site to the Fab region, this often results in reduced binding efficiency. Some pioneering methods involve covalently linking the linker to the affinity peptide via stable covalent bonds, which are then covalently attached to the antibody to form ADCs.
–04–
3. Site-Specific and Site-Selective Conjugation
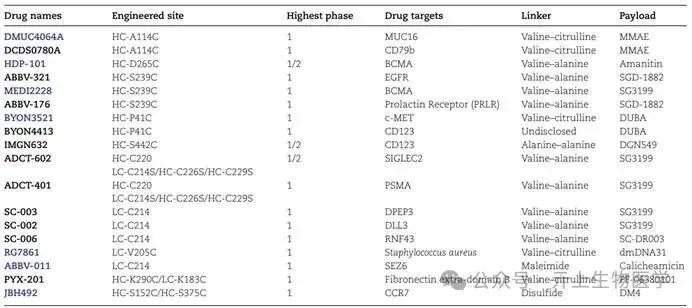
Engineered Cysteine random cysteine conjugation and re-bridging are techniques that utilize naturally occurring cysteine residues within the antibody structure. Unlike them, thiomab technology achieves selective and uniform modification at desired sites on the antibody by utilizing engineered reactive cysteine that does not involve structural disulfide bonds. Generally, the design of cysteine mutations aims to facilitate the coupling of cytotoxic payloads while maintaining the stability, affinity, and minimizing ADC aggregation of monoclonal antibodies. To determine the optimal positions for mutations, several techniques are typically employed, including computational modeling, model system screening, and high-throughput scanning.
Junutula et al. first reported a thiomab strategy, replacing the alanine (HC-A114C) at position 114 of the heavy chain of the anti-MUC16 antibody with engineered cysteine residues, allowing the reactive thiol within the engineered position to react with maleimide-loaded linkers. The synthesized anti-MUC16 ADC demonstrated efficacy in xenograft mouse models and showed high-dose tolerance in rats and cynomolgus monkeys, establishing a general approach for thiomab conjugation strategies.
The table below details the engineered cysteine sites used in clinical stage ADCs.
Non-Standard Amino Acids
In addition to thiomab technology, the incorporation of non-standard amino acids (ncAA) provides another possibility for site-specific conjugation. This technology uses amino acids with unique chemical structures, allowing for the chemical selective introduction of linker-payload complexes. This technology requires the re-engineering of antibody sequences, utilizing orthogonal tRNAs and aminoacyl-tRNA synthetases (aaRS) that are orthogonal to all endogenous tRNAs and synthetases in the host cell, to incorporate ncAA into proteins in response to unassigned codons. Typically, ncAA is added to the culture medium during fermentation. Choosing non-natural amino acids is crucial, as they may elicit immunogenicity. Commonly used ncAA are analogs of natural amino acids with unique groups, such as ketones, azides, cyclopropenes, or dienes.
Studies have successfully integrated para-acetylphenylalanine (pAcF) into the anti-CXCR4 antibody. The payload Auristin was effectively conjugated to the antibody via oxime linkage, generating a chemically homogeneous ADC. This ADC demonstrated good in vitro activity and completely cleared lung tumors in mice.
Due to the acidic conditions required for oxime linkage and the slow release kinetics of ADCs, another option is to incorporate azides containing ncAA. The widely used para-azidophenylalanine (pAzF) can rapidly undergo CuAAC or SPAAC reactions under physiological conditions, successfully conjugating glucocorticoid payloads to the anti-CD74 antibody. In addition to pAcF technology, azide-containing lysine analogs (AzK) have also been successfully integrated into antibodies to produce site-specific ADCs with Auristin, PBD dimers, or tubulin payloads.
Moreover, cyclopropene derivatives of lysine (CypK) and naturally occurring atypical amino acids, such as selenocysteine (Sec), have been successfully integrated into antibodies. The resulting ADCs exhibit good stability, selectivity, and in vitro and in vivo activity.
–05–
Conclusion
Due to the broad impact of conjugation technologies on ADC characteristics, determining the optimal conjugation sites and chemistries for specific antibody/linker/payload combinations remains a significant challenge. These impacts include binding, internalization, payload release, PK, effector functions, and more. Although some emerging conjugation technologies often demonstrate better efficacy and safety, there is frequently a gap in translation between preclinical and clinical studies. Furthermore, even if the entire process appears simpler, new conjugation technologies can introduce unexpected challenges in the CMC process. As our understanding of the complexities of ADCs deepens, and as we accumulate clinical data from more ADCs employing advanced conjugation methods, new and more suitable conjugation technologies are expected to emerge. These technological advancements will further address unmet clinical needs.
References:
1. The Chemistry Behind ADCs. Pharmaceuticals (Basel). 2021 May; 14(5): 442
2. A review of conjugation technologies for antibody drug conjugates. Antib Ther. 2025 Apr 17; 8(2):157-170.
This public account contains articles that are intended to convey more information, and we clearly state the source and authors. We do not wish to be reposted by any media or individuals. If you have any questions, please contact us (shenyini@qishangbiomed), and we will promptly take action. All articles only represent the author’s views, and do not represent the position of this site.
