
Please click the “Bio New Thinking” above to follow for more exciting content!
Introduction
With the continuous improvement of biopharmaceutical research and clinical research levels, the research and application of Antibody-Drug Conjugates (ADCs) have been increasing. ADCs are mainly composed of antibodies, linkers, and small molecule toxins, combining high targeting specificity with high cytotoxicity. However, due to their complex and diverse structures and the low content of small molecule toxins released in the circulation system, pharmacokinetic studies related to ADCs face significant difficulties. Therefore, this article introduces the key considerations related to the pharmacokinetics of ADCs from the perspectives of molecular design ideas, pharmacokinetics and research strategies, and optimization strategies for ADCs.【Written by / Lone Poison Pharmacist】
Key Points
☀ Key considerations in ADC molecular design: targeting antibodies, small molecule toxins, linkers, and drug-antibody conjugation ratios /
☀ PK/PD characteristics of ADCs and their research strategies /
☀ ADC optimization strategies based on PK/PD: antigens, antibodies, linkers, small molecule toxins, drug resistance, tumor penetration, surface modifications, etc. /
Main Discussion
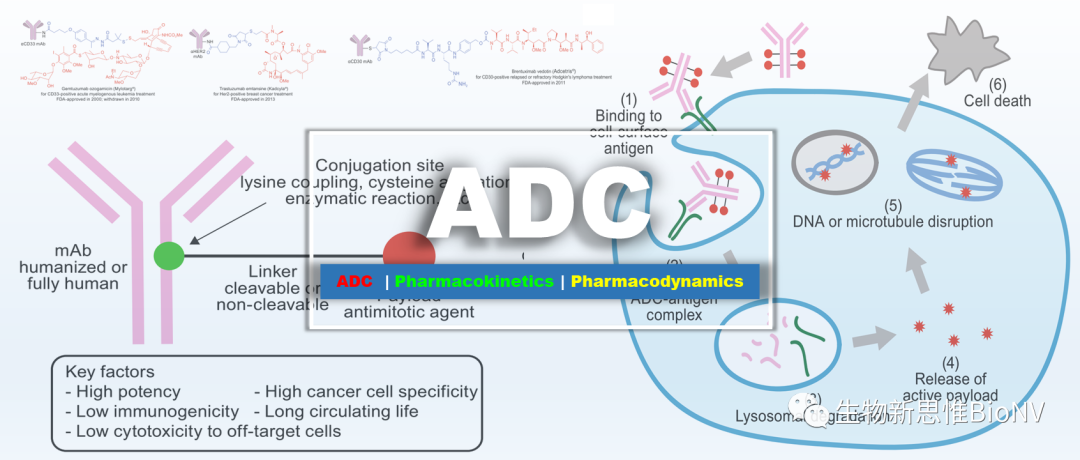
ADC drugs (Antibody-Drug Conjugate) refer to drugs prepared by coupling biologically active small molecule toxins (Payload) to monoclonal antibodies (Antibody) through linkers. These drugs deliver small molecule toxins specifically to tumor cells via antibodies that target tumor cell surface antigens, thereby killing tumor cells. Therefore, they possess the characteristics of specific binding between antibodies and target antigens, as well as high cytotoxic effects on tumor cells.
However, the complex molecular design of ADCs also brings many challenges to their drug development. Issues such as immunogenicity, low internalization rate, and instability of the linker may affect the safety and efficacy of the drugs. Different types of ADC molecules may vary significantly, even among ADC drugs targeting the same antigen, as the antigen epitopes, linker sites, linkers, and small molecule toxins of different molecules may differ, leading to variations in plasma stability, metabolic processes, pharmacokinetics/pharmacodynamics (PK/PD) relationships, and adverse drug reactions. Therefore, to obtain ADC drugs with ideal PK/PD characteristics, efficacy, and safety, it is crucial to consider the components and combination strategies of antibodies, linkers, and small molecule toxins, and to optimize them based on PK/PD.
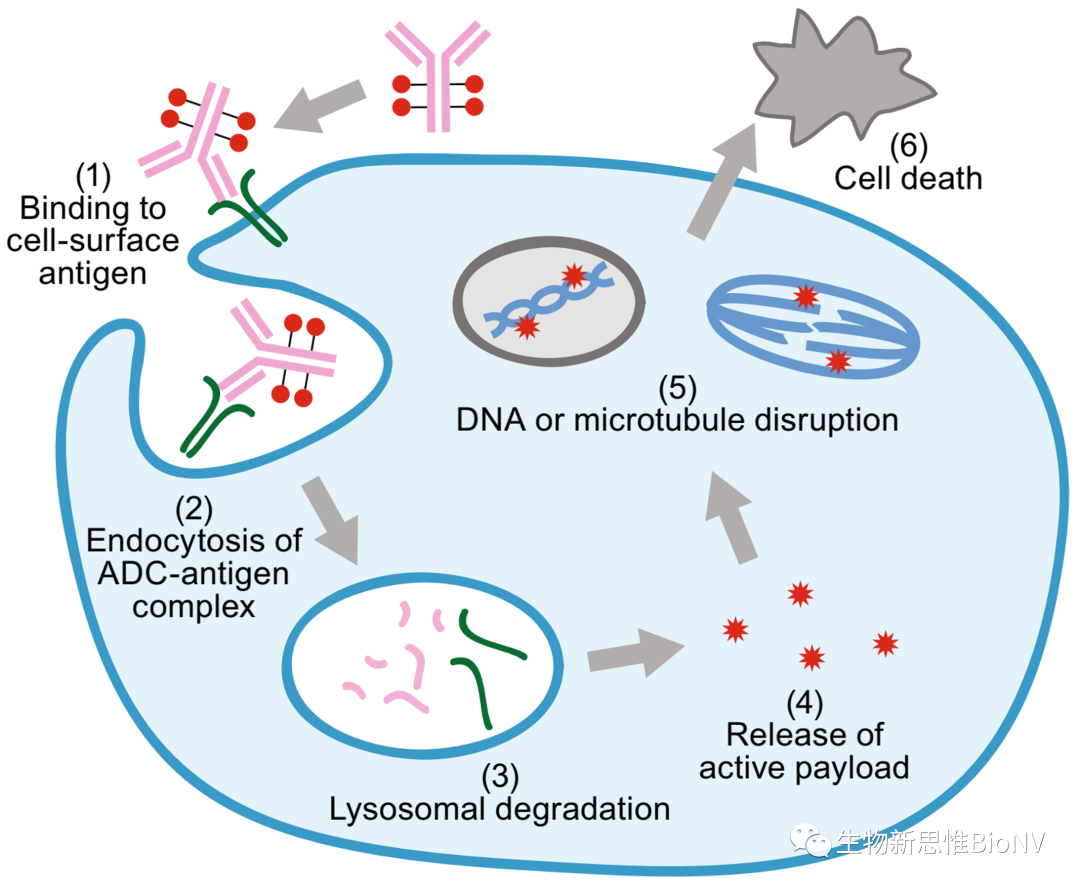
Figure 1 Mechanism of Action of ADC Drugs
 Key Considerations in ADC Molecular Design
Key Considerations in ADC Molecular Design
ADC molecules mainly consist of three parts: targeting antibodies, linkers, and small molecule toxins. The targeting antibody must efficiently and specifically bind to target cells; the linker must be stable enough and should be able to rapidly release the small molecule toxin in an active form after entering the target cell; the small molecule toxin should have a high killing effect on tumor cells. Generally speaking, all three components can affect the efficacy and safety of ADCs. For detailed structures of ADCs, please refer to“General Process and Key Considerations in ADC Development”, this article briefly elaborates on the main considerations in the molecular design of ADCs regarding these three components.
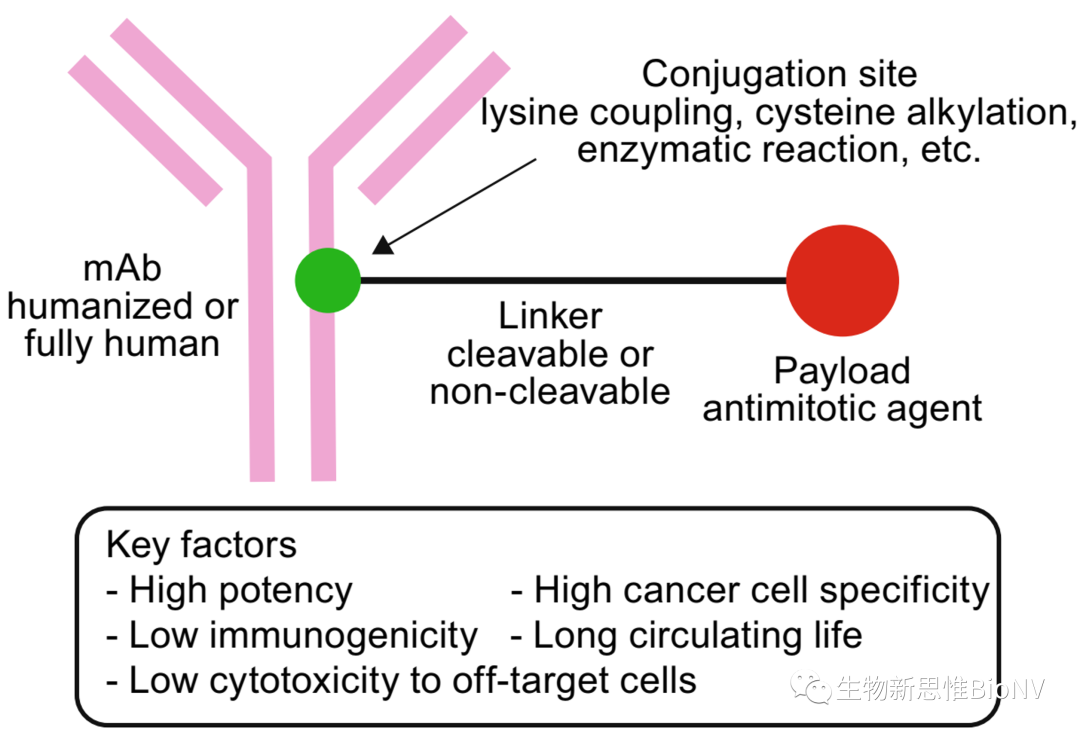
Figure 2 Molecular Structure and Key Indicators of ADC Drugs
Targeting Antibody (Antibody)
The targeting antibody is the decisive factor for the drug’s indications and is the starting point for ADC drug design. After determining the target indication, it is essential to identify which antigens are expressed at high levels on the surface of target cells, and then design targeting antibodies based on these antigens. The target antigens on the surface of target cells should have the following characteristics:
Antigens should generally be non-secretory /
Should release small molecule toxins through enzymatic degradation inside /
Antigens should be highly uniformly expressed on the surface of target cells /
Should have corresponding internalization pathways, and suitable internalization rates /
Antigens should not be expressed or expressed at low levels on normal tissues or cell surfaces /
Low expression of target antigens and low internalization rates are common issues in ADC drug design, which may lead to insufficient efficacy and off-target toxicity, posing significant challenges in ADC design. There are some strategies to address these issues, commonly including the following two:
Using anti-tumor angiogenesis antibodies: This can effectively avoid the internalization process but may lead to off-target effects, thus requiring careful selection of target antigens and corresponding antibodies /
Designing non-internalizing ADCs based on bispecific antibodies: Using bispecific antibodies to target two non-overlapping epitopes of one antigen to enhance the affinity between the antibody and the antigen /
Common antibodies used in ADC design are immunoglobulins (IgG), mainly IgG1, IgG2, and IgG4. IgG3 has a lower binding rate to FcRn receptors and is usually cleared faster, thus is not commonly used. IgG1 can further enhance ADC activity through ADCC and CDC, making it the most widely used subtype. However, the antibody molecule itself may affect the targeting of ADCs, reducing drug accumulation in target cells, shortening drug half-life, and even hindering drug molecules from entering target cells, so antibodies of appropriate molecular weight should be selected. The antibodies chosen in ADC design should have the following characteristics:
Have low immunogenicity /
Can maintain all or part of the function of the naked antibody /
Can efficiently deliver small molecule drugs to target cells /
Must have suitable sites on the antibody molecule for coupling with the linker /
The target cells must have corresponding internalization pathways for the antibody, with suitable internalization rates /
Small Molecule Toxins (Payload)
The expression levels of target cell surface antigens, the tumor penetration ability of antibodies, and internalization efficiency may become limiting factors for ADC drugs to enter target cells, leading to low intracellular concentrations of small molecule toxins. Therefore, small molecule toxins need to possess high cytotoxicity. Typically, the targets of small molecule toxins are located inside the cell; if ADCs cannot be transported into the cells, the efficacy and safety of the drugs will be affected. Conversely, if they dissociate outside the cell, they may be toxic to surrounding normal cells. In addition, small molecule toxins may also affect the overall properties of ADC drugs, such as internalization efficiency, polarity, immunogenicity, etc., so the molecular structure and its compatibility with the overall structure and function of ADC should be considered. Finally, small molecule toxins should have appropriate solubility and stability. Common small molecule toxins include: maytansine, auristatins, anthracyclines, and camptothecin analogs.
Linkers
The selection of linkers is crucial, as they can significantly impact the metabolic pathways of the drugs in vivo. On one hand, they should be stable enough to exist in the circulation system; on the other hand, they should be able to efficiently release small molecule toxins inside the target cells. Additionally, the molecular weight, structure, and polarity of the linker should be considered for their effects on the overall properties of ADCs. Linkers can be classified into cleavable and non-cleavable types, each with its advantages and disadvantages: non-cleavable linkers are more stable, can reduce off-target toxicity, and improve multidrug resistance (MDR); cleavable linkers produce metabolites that can enter cells through passive diffusion, generating bystander killing effects, which is important for tumors with heterogeneous expression of target antigens, but they are more prone to off-target effects.
Drug-Antibody Conjugation Ratio (DAR)
In addition to the three aforementioned factors, the efficacy of ADCs is also highly related to the drug-antibody conjugation ratio (DAR). Generally, improving DAR can increase the concentration of small molecule toxins in target cells. However, a higher DAR does not always equate to better efficacy; often, a higher DAR results in greater toxicity to normal tissues. Therefore, selecting an appropriate DAR is significant. Furthermore, the conjugation sites are related to the uniformity of the ADC, which is also an important consideration in DAR design. Cysteine and lysine residues on antibodies are more likely to undergo chemical reactions and modifications, making them common conjugation sites in ADCs. Each antibody can have up to 80 lysine residues, so using them as conjugation sites may increase the heterogeneity of ADCs. In contrast, each antibody contains only 8 free cysteines, making them advantageous for reducing the heterogeneity of ADCs.
PK/PD Characteristics of ADCs and Their Research Strategies
PK/PD Characteristics of ADCs and Their Influencing Factors
The main component of the ADC molecular structure is the targeting antibody; thus, ADC drugs exhibit pharmacokinetic characteristics and mechanisms similar to naked antibodies, such as target-mediated drug clearance, FcRn receptor recycling, and non-specific proteolytic degradation. Overall, similar to antibody drugs, the administration method of ADCs is intravenous injection, and their distribution characteristics are similar to those of antibody drugs, with metabolic and clearance pathways for both antibodies and small molecules, showing non-linear behavior at low doses and linear behavior at high doses. As mentioned earlier, the complex and diverse structures of ADCs, as well as the number of small molecule toxins conjugated to antibodies and the conjugation sites, lead to structural differences, making ADCs essentially a mixture of different molecules.
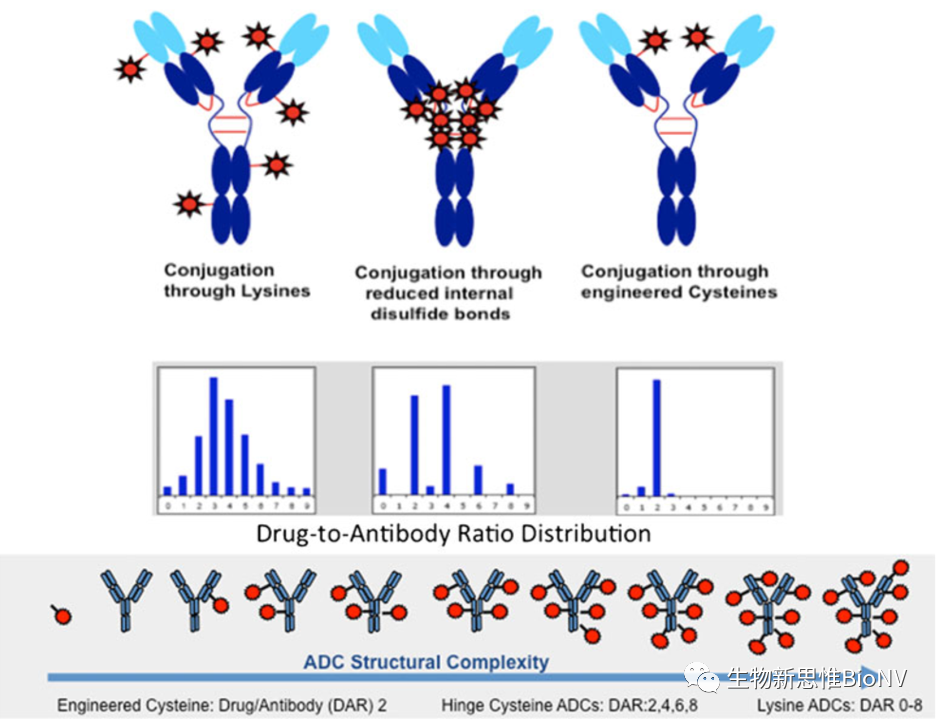
Figure 3 Structural Diversity of ADCs
Moreover, after entering the body, small molecule toxins will gradually dissociate from the ADC molecules, further increasing the diversity of ADCs. Overall, as shown in Figure 4, this continuously changing diversity and the intracellular and extracellular environments complicate the factors affecting the pharmacokinetics of ADCs, which is also a significant challenge in the PK research of ADC drugs.
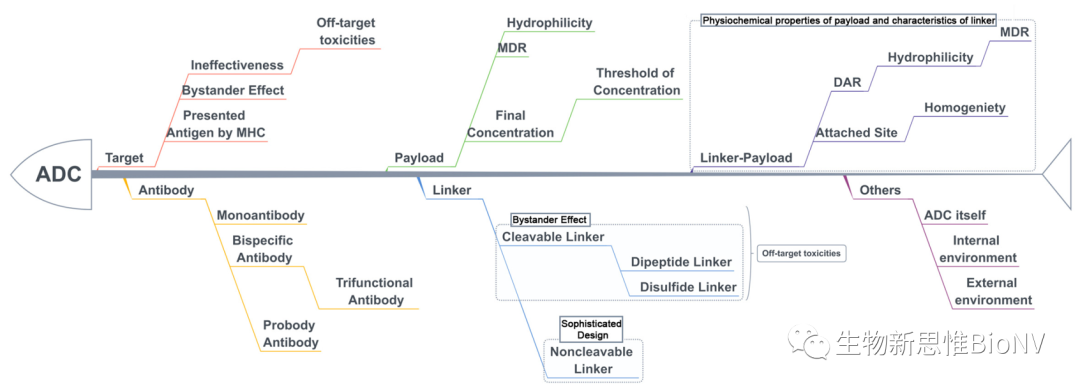
Figure 4 Key Influencing Factors of ADC Efficacy and Toxicity
PK/PD Research of ADC Drugs
Contents of PK/PD Research
The main contents of PK/PD research for ADC drugs include: stability of ADC molecules, blood concentration-time curves, distribution, metabolism, and excretion processes (ADME). If the small molecule toxin is a new compound, comprehensive in vivo and in vitro research methods should be considered, combined with qualitative and/or quantitative detection methods to study the systemic exposure, plasma protein binding, and uptake/distribution characteristics of small molecule toxins in tumors and normal tissues in detail. If necessary, the metabolic profile of its metabolites and the systemic exposure, distribution, release methods, and breakpoints of each important metabolite should also be studied. Generally, systematic studies should be conducted on the PK/PD characteristics of conjugated antibodies, total antibodies, conjugated effect molecules, free small molecule toxins, and their analogs, to characterize the overall PK/PD characteristics of ADCs.
In vivo, there are two clearance pathways for ADCs: one is the antibody part undergoing disintegration through enzymatic degradation, and the other is the complete dissociation of small molecule toxins from the antibody. Comparing the clearance rates of total antibodies and conjugated antibodies after ADC administration can reflect the speed at which small molecule toxins completely dissociate from ADC drugs. To evaluate the impact of conjugated drugs on antibody metabolism, the total antibody PK measured for naked antibodies can be compared with that of ADC drugs to evaluate the influence of small molecule toxins on the clearance rate of antibodies after being linked to them.
ADCs can also induce immune responses that produce anti-drug antibodies (ADA), with both product-related intrinsic factors and body-related extrinsic factors potentially influencing the incidence of ADA. The ADA produced in the body can neutralize ADCs, accelerating the clearance rate of ADCs and naked antibodies in the body. Therefore, strict monitoring and evaluation of the immunogenicity of ADCs are necessary in clinical trials.
PK/PD Research Strategies
Generally, during the design process of ADC drugs, PK/PD modeling methods can be used to quantitatively reflect the relationship between drug dosage and pharmacological effects, exploring suitable administration doses, frequencies, and dosage adjustment strategies through comprehensive assessment of exposure-response (ER) relationships. Compared to naked antibodies, ADCs have a narrower therapeutic index, thus requiring more refined ER analyses to support clinical research and practical usage. Due to the complexity of ADC’s own structure, its PK/PD analysis poses significant challenges, with a vast array of analytical indicators, making the establishment of ER relationships even more complex. Therefore, it is necessary to combine various methods, including in vitro, in vivo, animal, and human studies, to investigate the metabolic mechanisms and metabolites of ADCs. Specifically, through reasonable in vitro and in vivo research models, using target cells to conduct metabolic studies, or conducting cross-species plasma stability studies, the metabolic pathways of ADCs can be dissected, metabolites can be identified, and the relevance of preclinical species can be established, providing references for human clinical trials.
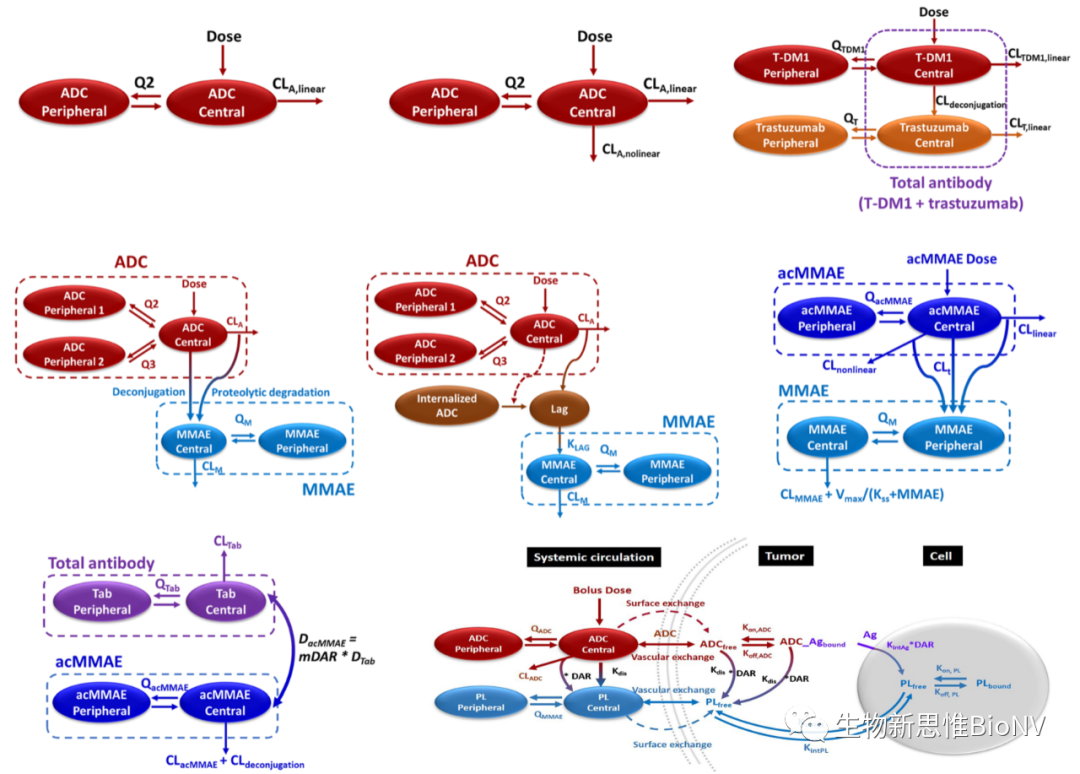
Figure 5 PK/PD Research Models of ADCs
Optimizing ADC Drugs Based on PK/PD
Through comprehensive and systematic PK/PD research, the biochemical, immunological, pharmacological, and molecular properties of ADCs can be studied in more detail, understanding their molecular characteristics, metabolic features, therapeutic mechanisms, and safety, thereby guiding future ADC drug design. The complexity and heterogeneity of ADCs often negatively impact their efficacy and therapeutic window, limiting their development in clinical applications and even leading to clinical trial failures. To overcome these issues, the following optimization strategies can be employed.
Optimizing Antigens
As mentioned earlier, the specificity, expression levels, internalization efficiency, and binding and dissociation efficiency of antigens with small molecule toxins are important characteristics affecting ADC efficacy and safety and are also crucial directions for antigen optimization. In addition, the homogeneity of target antigen expression in tumor types and positive patients also significantly influences ADC efficacy, and antigen shedding or secretion may reduce the binding ability of ADCs to targets and increase toxicity risks. Therefore, optimizing these factors is crucial for antigen selection. Furthermore, identifying new target antigens is also an important avenue for improving ADC design, allowing for the design of new ADC molecules targeting new antigens, thus developing more effective and safer therapeutic methods.
Optimizing Antibodies
In ADC development, enhancing specificity, affinity, and pharmacokinetics-related strategies are crucial for optimizing their monoclonal antibody components. Over the past decade, the industry has developed methods using new bispecific antibodies and half-antibody technologies to enhance ADC efficacy. Studies have shown that bispecific and half-antibodies perform better than first, second, and third-generation ADCs based on monoclonal antibodies. Therefore, future searches for more new bispecific and half-antibodies can be conducted to design ADC drugs that can bind multiple antigens simultaneously.
Optimizing Linkers
The molecular structure and properties of linkers play a vital role in the efficacy, pharmacokinetics/pharmacodynamics, and therapeutic index of ADCs. In vitro studies have shown that ADCs designed with non-polar or non-charged linkers exhibit lower potency in cells. Therefore, developing hydrophilic or charged linkers by reducing hydrophobicity can enhance ADC potency. Moreover, several new linkers are currently being tested in preclinical trials, such as triamine peptide linker CX, pseudo-peptide linker cBu-Cit, and branched linkers.
New Small Molecule Toxins
Pyrrolobenzodiazepine (PBD) compounds are a new type of ADC linker derived from natural products with antibacterial and anticancer properties produced by several actinomycetes. They are sequence-selective DNA alkylating compounds that can kill cancer cells by binding and cross-linking to specific targets on cancer cell DNA. Thus, they can prevent tumor cell proliferation without altering the DNA helix, potentially avoiding the development of drug resistance. Additionally, substances such as calicheamicin, spliceostatin C (an RNA splicing inhibitor), and nitric oxide (NO) can also serve as small molecule toxins. In the future, new small molecule toxins can be designed to create more effective and safer ADCs, potentially overcoming the challenges of drug resistance.
Overcoming ADC Drug Resistance
As the application of ADCs expands, the issue of drug resistance (MDR) has become a significant limiting factor for clinical success. Factors leading to the heterogeneity of resistance mechanisms include increased expression of MDR1 proteins, changes in microtubule components, downregulation of antigen expression, internalization and intracellular transport of antigen-ADC, and reduced drug release. First, as mentioned earlier, the MDR problem can be addressed by replacing it with new small molecule toxins. Second, the properties of linkers can be altered to combat MDR, as the MDR1 protein is less efficient in transporting hydrophilic compounds than hydrophobic ones, so increasing the hydrophilicity of linkers can alleviate MDR. Finally, modifying the linker-small molecule toxin conjugation structure can also improve MDR.
Enhancing ADC Tumor Penetration
The key factors affecting the effective delivery of ADCs are the accessibility of tumors and antigens. The penetration ability of antibodies into tumors is limited, resulting in low delivery of ADC drugs, thus requiring highly toxic small molecule toxins to achieve the goal of killing tumor cells. However, excessive toxicity may impact surrounding normal cells. Therefore, the industry has been striving to enhance the tumor penetration of ADCs. Reducing the molecular weight of ADCs can eliminate the binding site barrier effect, thereby improving their tumor penetration, for example: bispecific antibodies, nanobodies, affibodies, DARPins, etc. Additionally, designing anchor protein repeat proteins (DARPins), non-IgG scaffolds, and non-internalizing monoclonal antibody scaffolds can selectively deliver and cleave in the tumor microenvironment through disulfide bond linkers with small molecule toxins, thus enhancing ADC tumor penetration. The biodistribution characteristics of ADCs may also affect their penetration into tumors, so determining the biodistribution of ADCs is crucial. Based on the measurement results, the physical mechanisms can be explored to study their tissue permeation and diffusion characteristics to support ADC design. Furthermore, the vascular and lymphatic systems also participate in ADC delivery; the charge, affinity, binding affinity to FcRn, and the heterogeneity caused by different DARs of ADCs can also affect ADC tumor penetration, thus these factors can also be optimized.
Surface Modifications
To improve human tolerance to ADC drugs and expand their therapeutic window, it is often necessary to alter the structure of drug carriers and formulation. This can generally be achieved by modifying linkers or antibodies to reduce clearance rates; glycosylation and PEGylation are two of the most common modification techniques: glycosylation, which involves adding carbohydrates (glycans) to the amino acid side chains of proteins or antibodies, is a post-translational modification whose optimization depends on the quantity and location of glycosylation; PEGylation involves adding non-immunogenic PEG polymers to another biomolecule to overcome certain drawbacks, which can reduce the immunogenicity of drugs, improve solubility, and extend the retention time of drugs in the body, as well as enhance the solubility of linkers and prevent aggregation.
Thought Extension
Thanks to the continuous progress in therapeutic monoclonal antibodies and conjugation technology, the development of ADC drugs has made rapid advances, and the uniformity of ADC molecules has also increased. Based on diversified PK/PD modeling and research technologies, various conjugation strategies and small molecule cytotoxins continue to emerge, providing many new directions for improving the efficacy, safety, and tumor delivery efficiency of ADCs.
Whether as a single drug or in combination with other therapies, a deeper understanding of the mechanisms of action and toxicity influencing factors of ADCs is essential. To enhance their therapeutic index, the minimum effective dose must be reduced by improving the potency of small molecule cytotoxins, or the maximum tolerated dose must be increased by improving tumor selectivity. In the future, further synthesis and characterization of more uniform and stable ADCs should be pursued, along with drug chemical control of their macromolecular structures, which is crucial for the clinical success of the next generation of ADCs. Tools for protein structural characterization, such as mass spectrometry, can be used to better understand the biological transformation pathways and mechanisms of ADCs in vivo, and this knowledge will help establish early development standards for all ADC components (antibodies, small molecule toxins, and linkers). Because ADCs are essentially a mixture, it is necessary to consider both their biological components (mAbs) and small molecule components (small molecule toxins and linkers) in terms of product quality attributes. Therefore, advanced analytical techniques such as native/ion mobility mass spectrometry, two-dimensional liquid chromatography, and capillary electrophoresis should be employed in early developability assessments to comprehensively characterize the structure and properties of ADCs, thereby gaining insights into important structural features related to ADC function.
Moreover, recent ADC designs have adopted some natural cytotoxic substances that are usually highly effective but often intolerable due to their high toxicity in humans, requiring further optimization or the exploration of more novel cytotoxic drugs. Additionally, to enhance the tumor penetration of ADCs, the continued development of protein scaffolds (DARPins, nanobodies, single-chain variable fragments (scFv), peptide-drug conjugates), antibody-drug conjugates (ADDCs), Fab fusion proteins, probody drugs (Probody), and other alternative forms of monoclonal antibodies is essential. These new molecules are highly comparable to full monoclonal antibodies in terms of toxicity, efficacy, and pharmacokinetics, and may even be applicable to more different indications.
With the increasingly rigorous and refined research methods for ADC-related PK/PD models, the development of new ADC drugs will become more manageable, and the data collection capabilities for clinical trials and biological analyses will continue to improve, undoubtedly having a more significant and far-reaching impact on the successful development of ADCs.
Previous Recommendations
Upstream Technology | Simplifying CRISPR Editing Processes to Improve iPSC Monoclonal Efficiency
Upstream Technology | Six Major Host Expression Systems for Therapeutic Proteins
Upstream Technology | Screening Strategies for Stable High-Yield CHO Cells
Upstream Technology | The Mysterious Post-Translational Modifications of Therapeutic Proteins – Tyrosine Sulfation and Its Regulatory Strategies
Upstream Technology | Protein Hydrolysates and Their Applications in Biopharmaceutical Production
Upstream Technology | CMC Cornerstone – Cell Line Development Processes in Biologics Production
Upstream Technology | The Industrial Breakthrough Path of Adherent Cells
Upstream Technology | Bispecific Antibodies Prone to Aggregation, Continuous Processes Solve the Problem
Upstream Technology | Key Influencing Factors and Regulatory Strategies for Monoclonal Antibody Glycosylation Modifications
Upstream Technology | New Hope in the Post-COVID Era: HEK293 Cells and Their Key Upstream Technologies
Upstream Technology | Diversified Continuous Flow Processes Drive Transformation of Biopharmaceutical Costs
Upstream Technology | Detailed Explanation of the Reduction and Fragmentation Issues of Bispecific Antibodies
Upstream Technology | Continuous Flow Processes Mitigate the Intracellular Mechanisms of Bispecific Antibody Aggregation
Upstream Technology | Core Technologies and Key Steps in the Process of Producing Recombinant Proteins Based on Cell Culture
Upstream Technology | How to Break Down Lactate Accumulation? Detailed Explanation of the Driving Mechanisms and Regulatory Strategies for Lactate Consumption in CHO Cells
Upstream Technology | Comprehensive Introduction to the Challenges and Countermeasures in High Mannose Modification Regulation of Monoclonal Antibodies
Upstream Technology | Understanding the Deep Regulatory Mechanisms of pH on Monoclonal Antibody PQA
Upstream Technology | Does Large-Scale Process Perform Better? Perhaps Because of High Hydrostatic Pressure
Upstream Technology | Is “Aggregation” the Fate of High Expressers? How to Resolve the Conflict Between Protein Aggregation and Secretion Efficiency?
Upstream and Downstream Technology | Process “Insight”: PAT and Digital Twins in Continuous Production of Biologics
Upstream and Downstream Technology | How to Regulate Acid/Base Heterogeneity? Review of the Sources, Impacts, Analyses, and Regulatory Strategies of Charge Heterogeneity
Downstream Technology | Drawing on Strengths: Purification Strategies for Bispecific Antibodies Based on Mixed-Mode Chromatography
Downstream Technology | Differentiating Hydrophilic and Hydrophobic: Purification Technologies Based on Hydrophobic Interactions in Bispecific Antibody Production
Downstream Technology | Size Matters: Purification Techniques for Bispecific Antibodies Based on Molecular Size
Downstream Technology | Charge-Based Purification Techniques for Bispecific Antibodies
Downstream Technology | “The Four Great Detectives”: Affinity Chromatography Capture Techniques for Bispecific Antibodies
Downstream Technology | Comprehensive Analysis of the “Reduction Causes” and “Solutions” for Disulfide Bonds in Antibodies
Downstream Technology | Research Progress on Monoclonal Antibody Purification Processes and Their Commercial Production
Characterization Technology | Efficient Characterization Strategies for HCP in Therapeutic Protein Production
Characterization Technology | Instability and Forced Degradation Research Strategies for Fc Fusion Proteins
Characterization Technology | Molecular Size Heterogeneity of Fc Fusion Proteins and Their Characterization Strategies
Characterization Technology | Glycosylation Modifications of Fc Fusion Protein Molecules and Their Characterization Strategies
Characterization Technology | Characterization Techniques for the Primary Structure of Therapeutic Fc Fusion Proteins
Characterization Technology | Charge Heterogeneity Characterization of Therapeutic Fc Fusion Proteins
Characterization Technology | Characterization Techniques for the Primary Structure of Therapeutic Fc Fusion Proteins
Characterization Technology | Detailed Explanation of New Techniques for Characterizing Monoclonal Antibody Glycosylation Modifications
Formulation Technology | The Stabilizing Effect of Polysorbate on Biologics
Formulation Technology | General Process and Key Considerations for Biopharmaceutical Formulation Development
Formulation Technology | Challenges and Innovative Solutions in Biologics Container Development
Cell Therapy | Regulating NK Cell Metabolism to Enhance Tumor Killing Efficiency
Special Topic Analysis | New Bottles for Old Wine: Challenges and Strategies in Upgrading Traditional Biopharmaceutical Processes
Special Topic Analysis | The Cornerstone and Bottlenecks of mRNA Technology Platforms
Special Topic Analysis | Fc-Fusion: Therapeutic Proteins Based on Antibody Fc Fragments
Special Topic Analysis | Major Challenges and Countermeasures in GMP Production and Quality Supervision of Biopharmaceuticals
Special Topic Analysis | Quality Management in the R&D Process of Biopharmaceuticals
Special Topic Analysis | Principles of Buffer System Selection and Supplier Management in Biopharmaceutical Production
Special Topic Analysis | The Metabolic Mechanisms of Tumor Evasion from NK Cell “Chase”
Industry Dynamics | New Breakthroughs in COVID Drugs: Professor Bai Lu and Dr. Dou Yang from Tsinghua University School of Pharmacy Publish Series of Articles Reporting COVID Drug Development Achievements
Industry Dynamics | General Process and Key Considerations in ADC Development
Industry Dynamics | Who Will Disrupt the Future of Innovative Drugs in the Next 20 Years?
Industry Dynamics | Leading Companies and Their Pipelines in the mRNA Vaccine Field
References
1 . A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nature reviews Drug discovery, 2017, 16, 315-337.
2 . A. Palumbo, F. Hauler, P. Dziunycz, K. Schwager, A. Soltermann, F. Pretto, C. Alonso, G. Hofbauer, R. Boyle and D. Neri, British journal of cancer, 2011, 104, 1106-1115.
3 . A. T. Lucas, L. S. Price, A. N. Schorzman, M. Storrie, J. A. Piscitelli, J. Razo and W. C. Zamboni, Antibodies, 2018, 7.
4 . B. Hughes, Nature reviews. Drug discovery, 2010, 9, 665-667.
5 . B. Wei, J. Gunzner-Toste, H. Yao, T. Wang, J. Wang, Z. Xu, J. Chen, J. Wai, J. Nonomiya and S. P. Tsai, Journal of medicinal chemistry, 2018, 61, 989-1000.
6 . D. Gervais, Therapeutic Enzymes: Function and Clinical Implications, 2019, 55-80.
7 . F. Li, K. K. Emmerton, M. Jonas, X. Zhang, J. B. Miyamoto, J. R. Setter, N. D. Nicholas, N. M. Okeley, R. P. Lyon and D. R. Benjamin, Cancer research, 2016, 76, 2710-2719.
8 . F. Loganzo, X. Tan, M. Sung, G. Jin, J. S. Myers, E. Melamud, F. Wang, V. Diesl, M. T. Follettie and S. Musto, Molecular cancer therapeutics, 2015, 14, 952-963.
9 . F. Sun, Y. Wang, X. Luo, Z. Ma, Y. Xu, X. Zhang, T. Lv, Y. Zhang, M. Wang and Z. Huang, Cancer research, 2019, 79, 3395-3405.
10 . G. S. Hamilton, Biologicals : journal of the International Association of Biological Standardization, 2015, 43, 318-332.
11 . H. Donaghy, 2016.
12 . H. Merten, F. Brandl, A. Plückthun and U. Zangemeister-Wittke, Bioconjugate chemistry, 2015, 26, 2176-2185.
13 . H. Tang, Y. Liu, Z. Yu, M. Sun, L. Lin, W. Liu, Q. Han, M. Wei and Y. Jin, Frontiers in pharmacology, 2019, 10, 373.
14 . H. Zhou and F.-P. Theil, John Wiley & Sons, 2015.
15 . J. A. Hartley, Expert opinion on investigational drugs, 2011, 20, 733-744.
16 . J. Andreev, N. Thambi, A. E. Perez Bay, F. Delfino, J. Martin, M. P. Kelly, J. R. Kirshner, A. Rafique, A. Kunz and T. Nittoli, Molecular cancer therapeutics, 2017, 16, 681-693.
17 . J. Coffman, K. Bibbo, M. Brower, R. Forbes, N. Guros, B. Horowski, R. Lu, R. Mahajan, U. Patil and S. Rose, Biotechnology and bioengineering, 2021, 118, 3323-3333.
18 . K. Lin and J. Tibbitts, Pharmaceutical research, 2012, 29, 2354-2366.
19 . K. Tsuchikama and Z. An, Protein & cell, 2018, 9, 33-46.
20 . L. R. Saunders, A. J. Bankovich, W. C. Anderson, M. A. Aujay, S. Bheddah, K. Black, R. Desai, P. A. Escarpe, J. Hampl and A. Laysang, Science translational medicine, 2015, 7, 302ra136-302ra136.
21 . L. Shefet-Carasso and I. Benhar, Drug Resistance Updates, 2015, 18, 36-46.
22 . M. Damelin, W. Zhong, J. Myers and P. Sapra, Pharmaceutical research, 2015, 32, 3494-3507.
23 . M. P. Deonarain, G. Yahioglu, I. Stamati and J. Marklew, Expert opinion on drug discovery, 2015, 10, 463-481.
24 . M. R. Junttila and F. J. De Sauvage, Nature, 2013, 501, 346-354.
25 . M. Shibata-Koyama, S. Iida, H. Misaka, K. Mori, K. Yano, K. Shitara and M. Satoh, Experimental hematology, 2009, 37, 309-321.
26 . O. Ab, K. R. Whiteman, L. M. Bartle, X. Sun, R. Singh, D. Tavares, A. LaBelle, G. Payne, R. J. Lutz and J. Pinkas.
27 . P. Herbener, K. Schönfeld, M. König, M. Germer, J. M. Przyborski, K. Bernöster and J. Schüttrumpf, PloS one, 2018, 13, e0195823.
28 . P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone and M. L. Ulrich, Molecular Cancer Therapeutics, 2017, 16, 116-123.
29 . P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Molecular cancer research: MCR, 2020, 18, 3-19.
30 . P. Malik, C. Phipps, A. Edginton and J. Blay, Pharmaceutical research, 2017, 34, 2579-2595.
31 . P. Zuo, The AAPS journal, 2020, 22, 1-13.
32 . Q. Zhou, J. E. Stefano, C. Manning, J. Kyazike, B. Chen, D. A. Gianolio, A. Park, M. Busch, J. Bird and X. Zheng, Bioconjugate chemistry, 2014, 25, 510-520.
33 . R. Gébleux, S. Wulhfard, G. Casi and D. Neri, Molecular cancer therapeutics, 2015, 14, 2606-2612.
34 . R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. Ten Hoeve, H. M. Janssen, A. H. van Onzen and P. J. Hudson, Nature communications, 2018, 9, 1-11.
35 . R. Singh, Y. Y. Setiady, J. Ponte, Y. V. Kovtun, K. C. Lai, E. E. Hong, N. Fishkin, L. Dong, G. E. Jones and J. A. Coccia, Molecular cancer therapeutics, 2016, 15, 1311-1320.
36 . S. E. Sedykh, V. V. Prinz, V. N. Buneva and G. A. Nevinsky, Drug design, development and therapy, 2018, 12, 195.
37 . S. Golfier, C. Kopitz, A. Kahnert, I. Heisler, C. A. Schatz, B. Stelte-Ludwig, A. Mayer-Bartschmid, K. Unterschemmann, S. Bruder and L. Linden, Molecular cancer therapeutics, 2014, 13, 1537-1548.
38 . S. Puthenveetil, F. Loganzo, H. He, K. Dirico, M. Green, J. Teske, S. Musto, T. Clark, B. Rago and F. Koehn, Bioconjugate chemistry, 2016, 27, 1880-1888.
39 . S. Sau, H. O. Alsaab, S. K. Kashaw, K. Tatiparti and A. K. Iyer, Drug discovery today, 2017, 22, 1547-1556.
40 . T. Gefen, J. Vaya, S. Khatib, N. Harkevich, F. Artoul, E. D. Heller, J. Pitcovski and E. Aizenshtein, International Immunopharmacology, 2013, 15, 254-259.
41 . T. R. Tipton, A. Roghanian, R. J. Oldham, M. J. Carter, K. L. Cox, C. I. Mockridge, R. R. French, L. N. Dahal, P. J. Duriez and P. G. Hargreaves, Blood, 2015, 125, 1901-1909.
42 . W. D. Hedrich, T. E. Fandy, H. M. Ashour, H. Wang and H. E. Hassan, Clinical pharmacokinetics, 2018, 57, 687-703.
43 . Y. Anami, W. Xiong, X. Gui, M. Deng, C. C. Zhang, N. Zhang, Z. An and K. Tsuchikama, Organic & biomolecular chemistry, 2017, 15, 5635-5642.
44 . Ji Shuangmin, Wang Yuzhu and Yang Jinbo, Chinese Journal of Clinical Pharmacology, 2021, 37, 6.
45 . Yang Yuemei and Shen Beifen, International Journal of Pharmaceutical Research, 2014.

Scan to Follow BioNV
Bio New ThinkingRead more exciting content
Copyright Statement
Bio New Thinking is an open knowledge sharing platform focused on biopharmaceutical R&D knowledge. All articles in this public account are for learning, communication, and knowledge sharing purposes. If there is any infringement, please scan the QR code below to contact us for deletion. Reproduced articles only represent the author’s views and do not represent the position of this public account.

Scan for Communication/Submission/Consultation/Cooperation