In August 2024, the Department of Pharmacy, China Medical University, in collaboration with the Key Laboratory of Precision Diagnosis and Treatment of Gastrointestinal Tumors, the Liaoning Provincial Engineering Research Center for Tumor Immune Peptide Drugs, and the Liaoning Provincial Key Laboratory for the Development and Evaluation of Molecular Targeted Antitumor Drugs Xiaoyun Hu (First Author) and Journal of Pharmaceutical Analysis published a research paper online titled: snoRNAs: The promising targets for anti-tumor therapy.

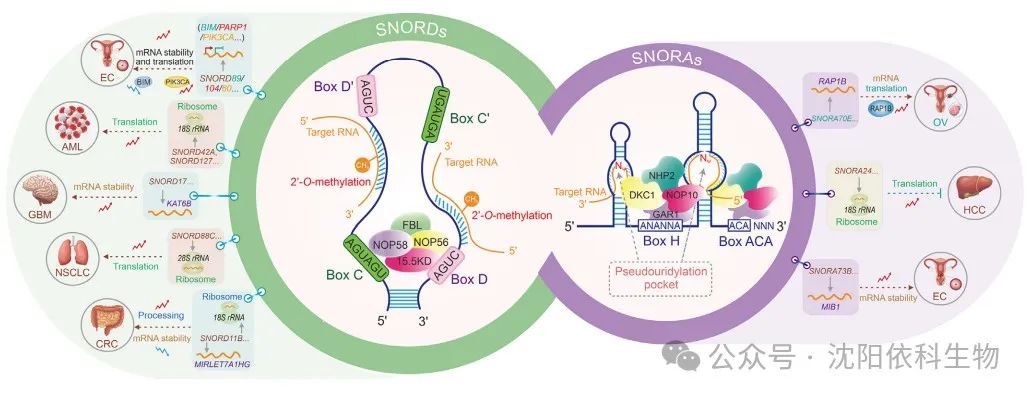
SnoRNA has traditionally been viewed as a housekeeping gene and often overlooked. However, the number of annotated snoRNAs in the human genome is steadily increasing. Eukaryotic snoRNAs are primarily located in the nucleus and typically range from 60 to 250 nucleotides in length. They can be divided into box C/D snoRNAs, responsible for 2′-O-ribose methylation, and box H/ACA snoRNAs, responsible for pseudouridylation. Additionally, there is a special subclass of snoRNA known as small Cajal body-specific RNAs (scaRNAs). scaRNAs regulate the 2′-O-ribose methylation and pseudouridylation of small nuclear RNAs (snRNAs) within the Cajal bodies in the nucleus. SnoRNAs mainly originate from protein-coding genes or introns of non-protein-coding genes, thus these genes are also referred to as snoRNA host genes (SNHGs). Subsequently, mature snoRNAs are cleaved and processed to produce metabolically stable snoRNA fragments, which are recognized as snoRNA-derived RNAs (sdRNAs), some of which exhibit characteristics similar to microRNAs (miRNAs). Furthermore, many snoRNAs that lack ribosomal RNA (rRNA) or snRNA recognition targets are labeled as ‘orphan snoRNAs’.
Based on secondary structure and conserved sequence motifs, snoRNAs can be divided into two classes: C/D box snoRNAs (SNORDs) and H/ACA box snoRNAs (SNORAs). SNORDs perform 2′-O-methylation at ribose sites, while SNORAs promote uridine isomerization. These classic modification mechanisms influence the folding and stability of rRNA, thereby affecting the occurrence and development of cancer. SnoRNAs dynamically interact with proteins to form small nucleolar ribonucleoprotein (snoRNP) complexes with catalytic activity, which are crucial for RNA synthesis, transport, and function. Each snoRNA family contains a unique set of binding proteins. The SNORD family includes four highly conserved core proteins: fibrillarin (FBL), nucleolar protein 56 (NOP56), NOP58, and 15.5KD. The binding proteins of SNORA, including dyskerin pseudouridine synthase 1 (DKC1), GAR1 ribonucleoprotein (GAR1), NHP2 ribonucleoprotein (NHP2), and nucleolar protein 10 (NOP10), are conserved and critical in the pseudouridylation process. More specifically, snoRNAs were initially thought to mediate the 2′-O-methylation and pseudouridylation of rRNA and snRNA. However, recent findings indicate that snoRNAs play significant roles in modifying messenger RNA (mRNA).
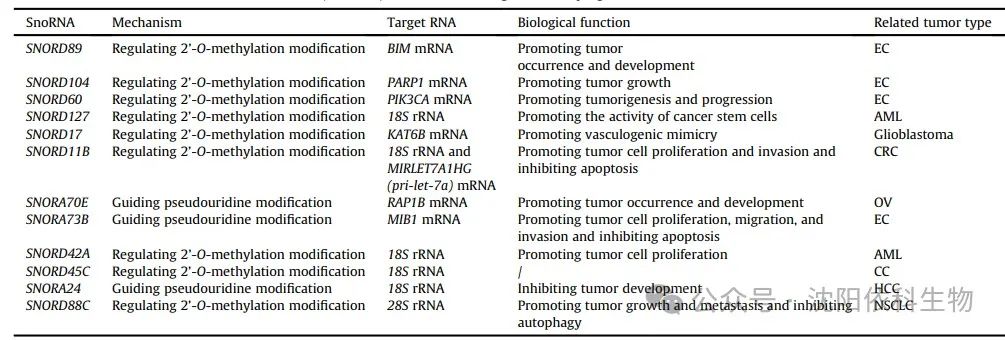
Most SNORDs guide 2′-O-methylation modifications at specific sites in rRNA through antisense complementarity. Antisense complementarity refers to the ability of a 10-21 nucleotide fragment upstream of the C/D box to form base pairs with the target RNA, leading to site-specific modifications. In endometrial cancer (EC) cells, SNORD89 expression is elevated and interacts with fibrillarin (FBL) to inhibit the translation of B-cell lymphoma-2 (BCL-2) homolog 11 (BIM) through 2′-O-methylation. This results in decreased BIM protein levels, thereby promoting EC cell proliferation and migration. SNORD104 binds to FBL and increases the stability and nuclear localization of poly(adenosine diphosphate (ADP)-ribose) polymerase 1 (PARP1) mRNA through 2′-O-methylation, thus promoting EC proliferation. SNORD60 interacts with FBL to enhance the stability and expression of PIK3CA mRNA through 2′-O-methylation, regulating the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway and promoting EC progression. Additionally, SNORD127 specifically recognizes the Gm1447 site on 18S rRNA, upregulating the 2′-O-methylation level of Gm1447, thereby promoting the activity of leukemia stem cells. In glioblastoma (GBM), SNORD17 promotes angiogenesis by modifying KAT6B through 2′-O-methylation, providing potential pathways for targeted therapy in clinical settings. In colorectal cancer (CRC), SNORD11B mediates the 2′-O-methylation at the G509 site on 18S rRNA, promoting the processing and maturation of 18S rRNA. Furthermore, SNORD11B also induces 2′-O-methylation modifications at the G225 site of MIRLET7A1HG (pri-let-7a). This modification leads to the degradation of pri-let-7a, reduced expression of the mature tumor suppressor let7a-5p, and upregulation of downstream oncogenes MYC (c-MYC) and RAS translation, thus promoting the occurrence of colorectal cancer.
SNORAs are crucial for pseudouridylation, a process that modifies specific sites of eukaryotic rRNA and snRNA. Pseudouridylation involves converting uridine in the target RNA molecule to pseudouridine. SNORA molecules possess a conserved secondary structure known as the ‘hairpin-loop-hairpin tail’. The guiding sequence of SNORA is located within one or two hairpins in this structure, forming a ‘pseudouridylation pocket’. This pocket facilitates non-canonical base pairing with the target uridine through interaction with precursor rRNA. In ovarian cancer (OV), SNORA70E interacts with keratin 17 to induce pseudouridylation of RAP1B mRNA, subsequently leading to increased levels of RAP1B protein and promoting the proliferation, invasion, and migration of OV cells. SNORA73B binds to DKC1 to enhance the stability and expression of MIB1 mRNA through pseudouridylation. This process leads to increased levels of Jagged 1 (JAG1) and activates the Notch pathway, ultimately promoting the development of EC.
Ribosomes are often referred to as the ‘protein production machines’, facilitating the dehydration condensation of amino acids to form peptide chains during translation. Therefore, the modification of rRNA, particularly 2′-O-methylation and pseudouridylation, contributes to the plasticity of ribosomal translation function, considering the evidence that these modifications regulate the translation program of cancer cells, which is a critical factor. For example, it has been shown that rRNA with 2′-O-methylation affects the ability of ribosomes to translate mRNA in HeLa cells. Reduced levels of pseudouridylation alter ribosomal activity, particularly affecting translation mediated by internal ribosome entry sites (IRES) in breast cancer (BRCA) cells and overall translation fidelity. Abnormal rRNA methylation regulated by the P53/FBL axis impairs ribosomal translation fidelity, activating IRES-dependent translation of oncogenes (such as IGF1R), thus promoting BRCA progression. In recent years, increasing evidence suggests that snoRNA-mediated classical modifications play important roles in ensuring ribosomal translation fidelity, potentially impacting the occurrence and development of cancer. SNORD42A guides 2′-O-methylation at the U116 site on 18S rRNA, promoting the translation of ribosomal proteins in acute myeloid leukemia (AML) and enhancing the proliferation and survival of leukemia cells. SNORD45C induces 2′-O-methylation modifications at the C174 site on 18S rRNA, reshaping mRNA translation in cervical cancer (CC) cells. SNORD127 mediates 2′-O-methylation at the G1447 site on 18S rRNA to promote the preferential translation of mRNA, driving the AML stem cell phenotype. SNORA24 guides the modification of pseudouridine on 18S rRNA to reduce the frequency of translation errors and stop codon read-through, thereby inhibiting the development of hepatocellular carcinoma (HCC) during RAS-induced senescence. Furthermore, SNORD88C promotes 2′-O-methylation at the C3680 site on 28S rRNA in non-small cell lung cancer (NSCLC), increasing the translation of downstream lipogenic enzyme stearoyl-CoA desaturase 1 (SCD1), thus promoting the proliferation and metastasis of NSCLC. In summary, the importance of snoRNA-mediated classical modifications in ribosomal translation is increasingly recognized, providing valuable insights into tumor regulatory patterns.
(Image source: Journal of Pharmaceutical Analysis)
(Image source: Journal of Pharmaceutical Analysis)
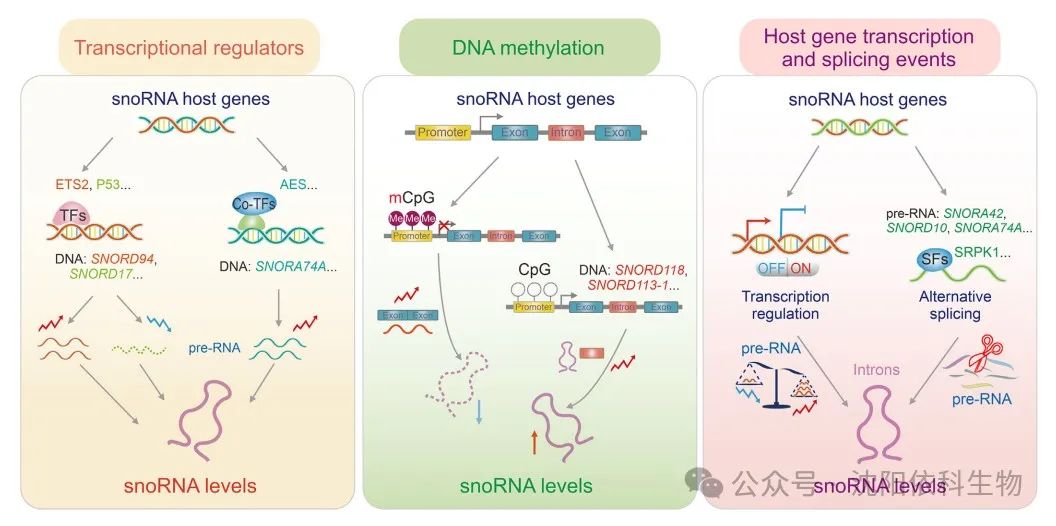
SnoRNAs, due to their conserved secondary structures and ability to form complexes with proteins and RNAs, have become metabolically stable products. Compared to mRNA (less than 6 hours), snoRNAs exhibit relatively long half-lives (24 to 48 hours), with their levels in cells regulated by both synthesis and degradation mechanisms. It is widely recognized that snoRNAs exhibit rich functional expression in various cancer types, stages, and metastasis, actively participating in tumor progression. Recent studies have shown that snoRNA expression can be regulated at the transcriptional level through interactions with transcriptional regulators and DNA methylation. Moreover, most snoRNAs are encoded within the introns of coding and non-coding genes, so their expression relies on the transcription and splicing events of host genes.
Increasing evidence suggests that transcription factors play key roles in regulating snoRNA expression, which is crucial for maintaining cellular homeostasis and ultimately affecting cancer progression. In mice with P53 mutations and osteosarcoma (OS), snoRNA is upregulated through transcriptional regulation dependent on E26 transformation-specific sequence 2 (ETS2), promoting tumor metastasis and suggesting that ETS2 may be a potential therapeutic target for P53 mutant osteosarcoma. P53, as a tumor suppressor, can inhibit the transcription of SNORD17, thereby suppressing the progression of hepatocellular carcinoma (HCC). Additionally, in acute myeloid leukemia 1-eighttwenty-one (AML1-ETO)-induced leukemogenesis, the amino-terminal split enhancer (AES), a co-transcription factor, has been found to influence various RNA processing pathways at the transcriptional level.
DNA methylation is a common epigenetic mechanism used to regulate gene expression. DNA methylation primarily occurs in the cytosine-phosphate-guanine (CpG) islands in the promoter regions of genes. High levels of CpG island methylation play significant roles in cancer induction. High-throughput sequencing results show that DNA methylation can inhibit the transcription of snoRNAs, thereby affecting cancer progression. In hepatocellular carcinoma (HCC) patients, the methylation level of the CpG island in the promoter region of the SNORD113-1 gene is significantly higher compared to adjacent non-tumor tissues, leading to a marked downregulation of its expression. SNORD113-1 exhibits tumor-suppressive properties by inhibiting the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and mothers against decapentaplegic homolog 2/3 (SMAD2/3) in the mitogen-activated protein kinase (MAPK/ERK) and transforming growth factor beta (TGF-β) pathways, thereby suppressing the proliferation of HepG2 and Huh7 cancer cells. Furthermore, a wealth of evidence suggests that highly methylated 5′-CpG islands are widely present in host genes, including SNORD123, U70C, and ACA59B, in colorectal cancer and lung cancer cell lines, leading to the epigenetic silencing of the associated snoRNAs.
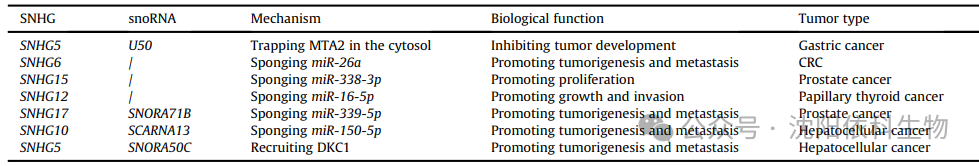
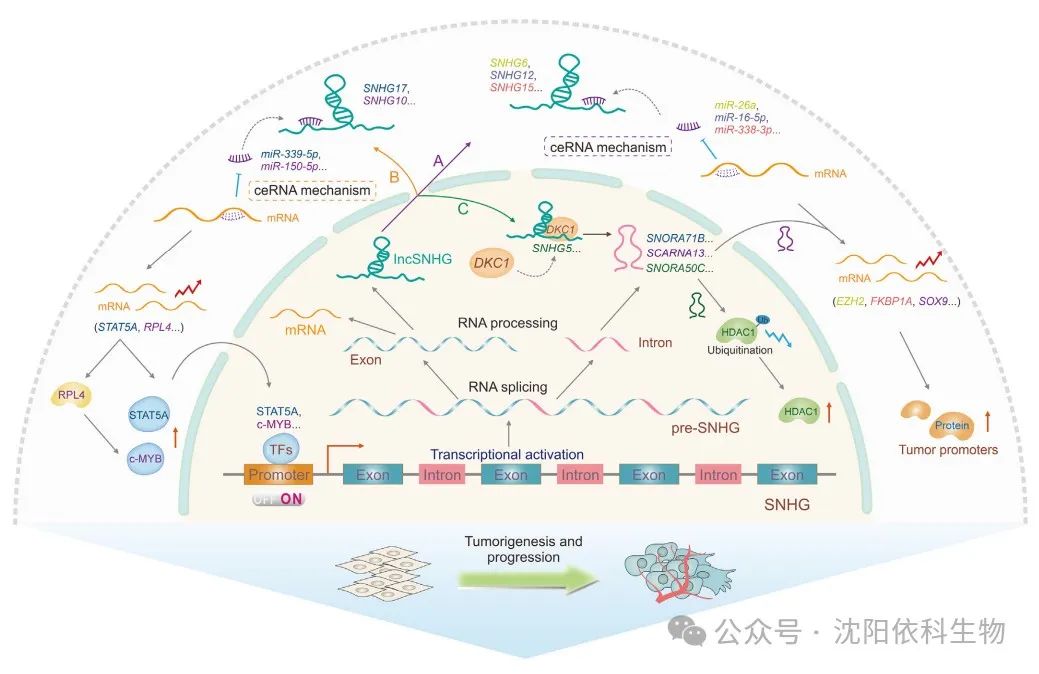
Recent studies indicate that most snoRNAs originate from the introns of coding or non-coding genes, and their expression depends on the transcription levels and splicing events of host genes. It has been discovered that snoRNAs embedded within the introns of protein-coding genes are produced through the transcription and splicing activities of their host genes. The host gene encodes full-length transcripts (also known as precursor RNAs) that include exons and introns. Within these transcripts, introns with nuclear localization are further processed into small molecules ranging from 65 to 300 nucleotides, thus defining them as snoRNAs. Therefore, the abundance of snoRNAs is directly dependent on the transcriptional regulation of their host genes. For instance, snoRNAs U14eU24 and E3 originate from SNHG1, which is actively transcribed into precursor RNA and releases through the splicing process that removes introns. Additionally, existing evidence suggests a co-expression relationship between intronic snoRNAs and their host genes, further confirming their transcriptional regulatory patterns. Moreover, the splicing patterns of host genes significantly affect the expression of their encoded snoRNAs. Specifically, genes encoding multiple snoRNAs often produce alternative transcript isoforms, allowing differential expression of individual co-encoded snoRNAs. A representative study confirmed that the splicing factor serine-arginine protein kinase 1 (SRPK1) upregulates the expression of SNORA42, SNORD10, and SNORA74A, which is associated with increased proliferation of gastric cancer cells.
(Image source: Journal of Pharmaceutical Analysis)
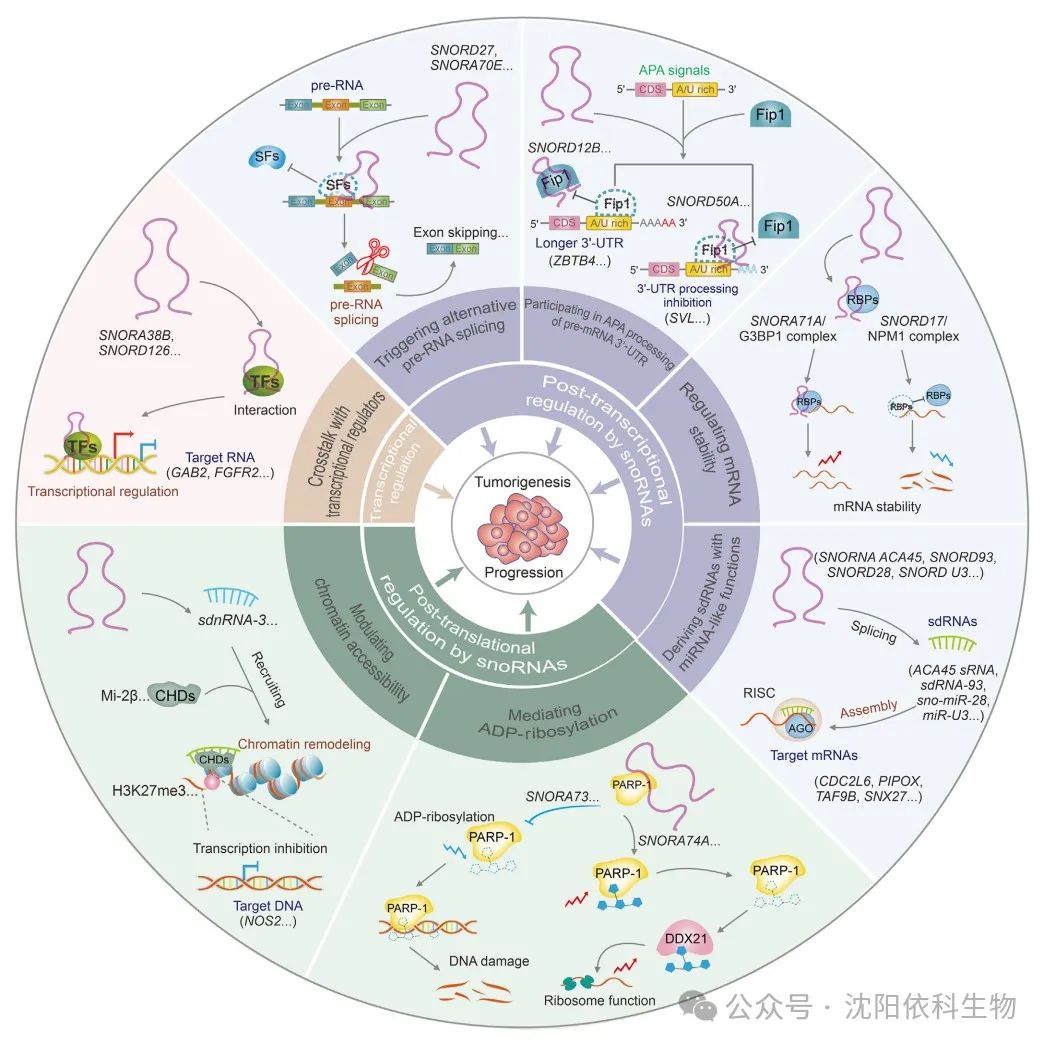
SnoRNAs play a significant role in the development and progression of tumors. In recent years, in addition to the two traditional mechanisms, novel regulatory mechanisms crucial for tumor occurrence and development have been discovered. The main regulatory mechanisms include transcriptional, post-transcriptional, and post-translational regulation of snoRNAs. Multiple studies indicate that snoRNAs can indirectly regulate tumor development and progression through selective modifications of target RNAs, while snoRNAs directly binding to target RNAs regulate their post-transcriptional modifications and contribute to post-translational regulation.
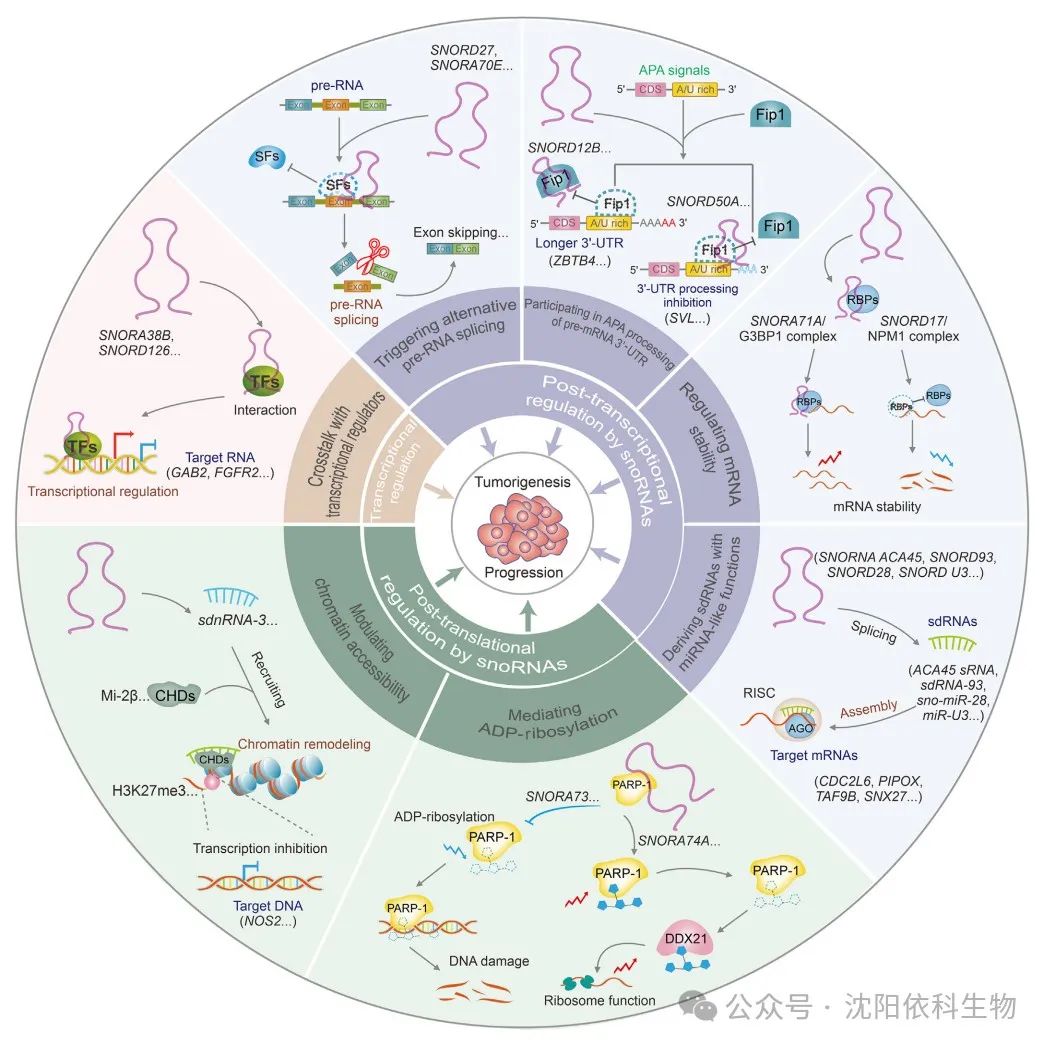
Interactions between snoRNAs and transcriptional regulators can modulate the expression levels of key cancer-related genes, thus triggering the development of malignancies. For example, in non-small cell lung cancer (NSCLC), SNORA38B promotes the transcription of the downstream GAB2 gene by directly interacting with the transcription factor E2F transcription factor 1 (E2F1), thereby activating the AKT/mTOR pathway and driving the progression of NSCLC. Additionally, SNORD126 can directly bind to heterogeneous nuclear ribonucleoprotein K (hnRNPK), leading to the upregulation of FGFR2 transcription and activation of the PI3K-AKT pathway, thus promoting the progression of hepatocellular carcinoma (HCC).
SnoRNAs are a type of snRNA molecule that plays a key role in regulating gene expression. Recent studies show that snoRNAs can cause various biological changes in tumors by affecting target splicing and RNA levels. SNORA70E is a splicing regulatory factor that influences the selective splicing of PARPBP in ovarian cancer (OV), leading to the exclusion of exon 4 in the PARPBP-88 transcript and the generation of a novel transcript PARPBP-15. This effect subsequently promotes the invasion, migration, and proliferation of OV cells. Similarly, SNORD27 competitively binds to splicing factors in HeLa cells, leading to the exclusion of exon 12 in the E2F7 precursor mRNA while inhibiting the inclusion of silenced exons in the precursor mRNAs of FER, ZBTB37, MAP4K3, and ABCA8, ultimately promoting cell proliferation. SNORD88C (also known as HBII-180C) regulates the splicing of fibroblast growth factor receptor 3 (FGFR3) mRNA, contributing to the development of various cancers. Additionally, a recent study indicated that SNORD75 can regulate the splicing of GAS precursor long non-coding RNA (lncRNA) by recruiting and binding to the methyltransferase-like 3 (METTL3)/METTL14 complex based on N6-methyladenosine (m6A) modification factors.
APA (alternative polyadenylation) is a key post-transcriptional regulatory mechanism during eukaryotic mRNA maturation, used for modifying and processing the 3′ end of precursor mRNA. SnoRNAs regulate APA modifications, thereby regulating mRNA 3′ end processing and affecting the expression of cancer-related genes. In glioblastoma (GBM) cells, SNORD12B is upregulated, and the binding of FIP1L1 (also known as FIP1) promotes the use of distal polyadenylation sites by the transcriptional repressor ZBTB4 during APA, resulting in an increased proportion of long 3′ UTR transcripts of ZBTB4, thereby downregulating ZBTB4 expression and ultimately inhibiting glucose and lipid metabolism as well as the proliferation of GBM cells. Additionally, SNORD50A acts as a direct APA regulatory factor, inhibiting the 3′ end processing of SVL mRNA in HeLa cells by disrupting the FIP1-PAS interaction.
Several studies indicate that snoRNAs can act as post-transcriptional regulatory factors, influencing mRNA stability and thus promoting cancer development. For instance, SNORA71A binds to the protein G3BP1 that regulates mRNA stability, increasing its stability and upregulating the negative regulatory factor of TGF-β, ROCK2, which promotes the growth and metastasis of breast cancer (BRCA). Upregulated SNORD17 binds to NPM1, reducing the stability of the tumor suppressor protein P53 and promoting the occurrence and metastasis of HCC. Therefore, snoRNAs play essential roles in cancer progression by regulating mRNA stability and influencing specific signaling pathways. Further in-depth studies will provide clearer insights to unravel the specific mechanisms by which these molecules impact various diseases.
Small nucleolar RNAs (snoRNAs) can produce small molecules that bind to argonaute (AGO) proteins, exhibiting functions similar to miRNAs. These small molecules are referred to as sdRNAs or snomiRNAs. Preliminary studies have shown that the nucleolar RNA ACA45 can produce stable sdRNA, namely ACA45 sRNA, in HEK293 cells. Similar to miRNAs, this molecule can bind complementarily to the 3′ untranslated region (3′ UTR) of CDC2L6 and inhibit its expression. sdRNAs can mimic the functions of miRNAs by fully or partially binding to target mRNAs, promoting gene silencing and affecting the occurrence and development of tumors. For example, sdRNA-93 (also known as HBII-336) derived from snoRNA93 exhibits miRNA-like characteristics. In breast cancer (BRCA) cells, this highly expressed sdRNA-93 can bind to the 3′ UTR of PIPOX and inhibit its expression, inducing invasive characteristics in specific BRCA cell types (such as MCF-7 and MDA-MB-231). Additionally, sno-miR-28 promotes the proliferation of BRCA cells by directly targeting the upregulated TAF9B 3′ UTR and indirectly decreasing P53 stability. Box C/D nucleolar RNA U3 is overexpressed in various cancers. In HCT116 cells, U3 nucleolar RNA is converted into miRNA-like miR-U3. This allows miR-U3 to bind to the 3′ UTR of SNX27 mRNA, potentially affecting various cellular functions. Similarly, miR-U3 also binds to the 3′ UTR of ZBTB4, reducing the stability of ZBTB7A mRNA. This leads to downregulation of ZBTB7A protein expression, promoting the glycolytic capacity and proliferation of glioblastoma cells. Targeting these functional nucleolar RNAs and their small molecular derivatives, such as using antisense oligonucleotides (ASOs) and small molecules, may be an effective strategy similar to inhibiting oncogenic miRNAs.
Nucleolar RNAs affect the post-translational modifications of nuclear proteins by influencing the catalytic activity of PARP1, which is significant for the occurrence and development of cancer. Recent studies indicate that the chromatin-associated orphan nucleolar RNA SNORA73 can bind to PARP1 to form nucleolar RNPs, inhibiting the auto-poly(ADP-ribosyl)ation of PARP1, thereby affecting the levels of DNA damage. Consequently, this process exacerbates genomic instability and promotes the progression of acute myeloid leukemia (AML). It has been found that in BRCA1/2 wild-type breast cancer (BRCA) cells, SNORA74A can activate the self-modification of PARP-1. This activation occurs through the interaction of PARP-1 with the RNA helicase DDX21, leading to site-specific ADP-ribosylation of DEAD-box helicase 21 (DDX21). Therefore, this process promotes the growth of breast cancer cells by enhancing ribosomal biogenesis and protein translation. To further understand the critical roles of these nucleolar RNAs in cancer progression, it is necessary to explore their regulatory mechanisms on PARP1 and DNA damage repair (DDR) pathways.
Nucleolar RNAs facilitate the selective recruitment and binding of chromatin-modifying factors to regulate chromatin accessibility. Nucleolar RNAs can interact with Drosophila decondensation factor 31 (DF31) to form ribonucleoprotein (RNP) complexes, leading to the decondensation of higher-order chromatin structures and the maintenance of high levels of chromatin structure. Therefore, nucleolar RNAs and their derivatives may influence cancer development through chromatin structural modifications. Bisphenol A promotes the development of prostate cancer by recruiting histone H3 lysine 9 trimethylation (H3K9me3), H3K4me3, and H3K27me3, thereby inhibiting the activity of SNORD59A, SNORD82, SNORD116, and SNORD117. Recent studies have identified dicer-independent small non-coding RNAs (sdnRNAs) derived from nucleolar RNAs (sdnRNA-3) and snoRNAs that inhibit the transcription of NOS2 in macrophages. This is achieved through the recruitment of the Mi-2b protein, thereby blocking the accessibility of the NOS2 gene promoter and promoting tumor growth.
(Image source: Journal of Pharmaceutical Analysis)
(Image source: Journal of Pharmaceutical Analysis)
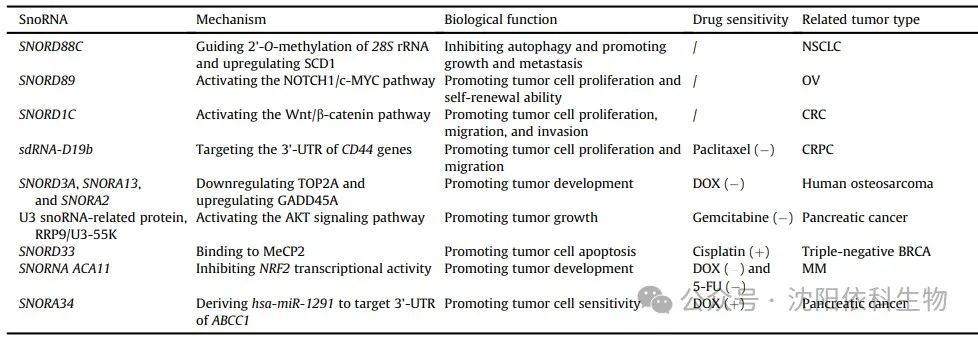
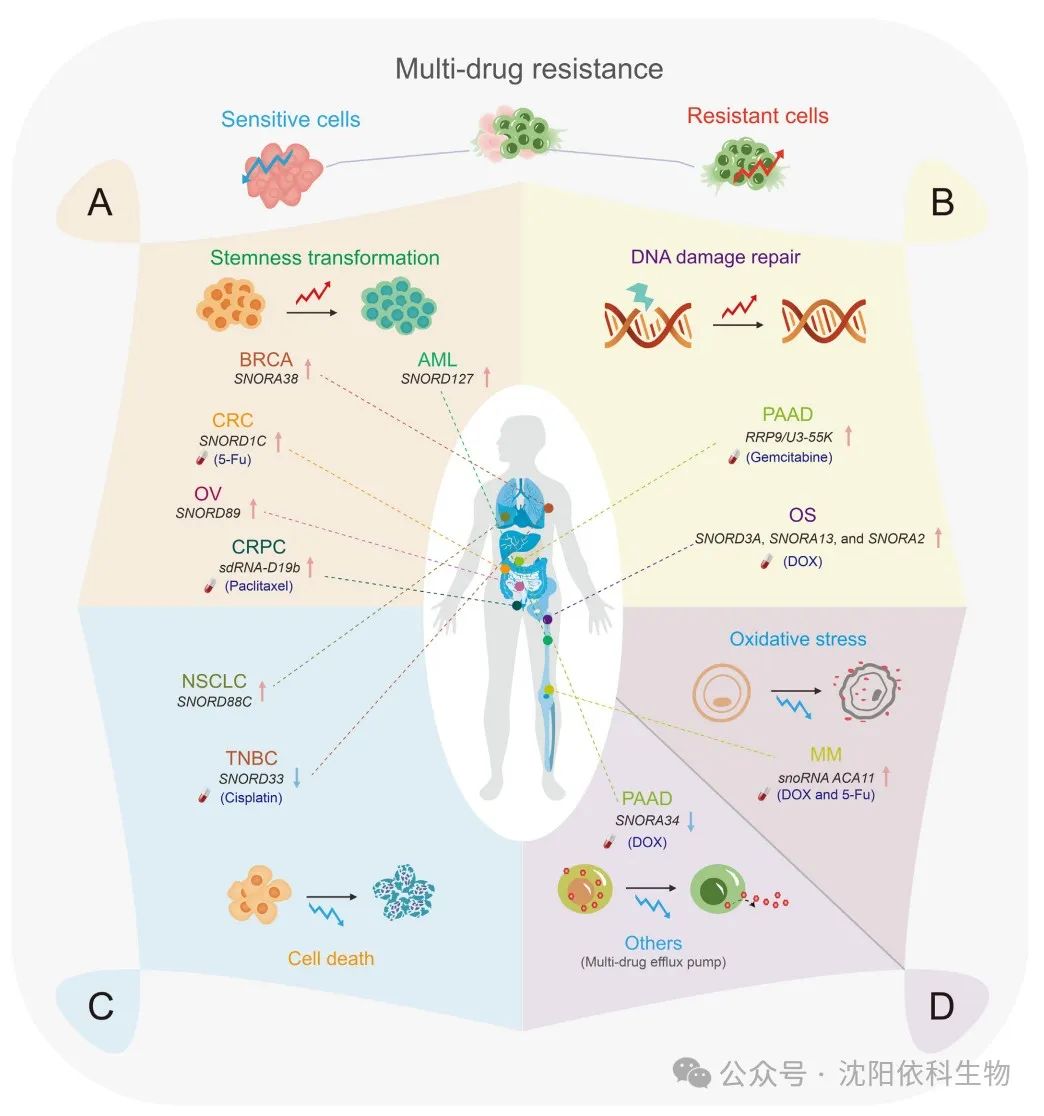
Numerous studies have demonstrated the regulatory role of snoRNAs in maintaining tumor cell stemness, which significantly affects tumor occurrence and development. Notably, SNORD127 triggers the preferential translation of amino acid transporter mRNA in a codon-dependent manner, driving the acute myeloid leukemia (AML) stem cell phenotype, such as self-renewal. Similarly, SNORD89 activates the NOTCH1/cell MYC (c-MYC) pathway and induces stemness in ovarian cancer (OV) cells. SNORA38 is positively correlated with the OCT-4 stem cell marker and regulates stemness in breast cancer (BRCA) cells. Furthermore, snoRNAs can transform tumor stem cells by modulating the expression of stem cell markers, leading to uncontrolled tumor proliferation and drug resistance. In colorectal cancer (CRC), the overexpression of SNORD1C activates the Wnt/β-catenin pathway, leading to the upregulation of CD44, SOX2, and other stem cell genes. This activation enhances the proliferation of colon cancer cells and cell cycle progression, promoting resistance to 5-fluorouracil (5-FU). Additionally, sdRNA-D19b selectively targets the 3′ untranslated region (3′ UTR) of CD44 mRNA, increasing the proliferation and metastasis of castration-resistant prostate cancer (CRPC) cells. This mechanism confers resistance to the drug paclitaxel. This study suggests that sdRNA may serve as a potential biomarker and therapeutic target for clinical intervention in CRPC.
Tumor cells possess robust DNA repair mechanisms that effectively repair DNA damage caused by chemotherapy, thereby enhancing drug resistance. In colorectal cancer cells, doxorubicin (Dox)-induced DNA damage leads to the expression of snoRNA derived from growth arrest-specific transcript 5 (GAS5) in a P53-dependent manner, indicating that snoRNAs may impact DNA damage. SnoRNAs and their associated proteins are involved in DNA damage response (DDR), which has significant implications for drug resistance. In Dox-resistant osteosarcoma cells, the upregulation of SNORD3A, SNORA13, and SNORA2 is crucial for synthesizing Dox-resistant proteins by downregulating the Dox target DNA topoisomerase IIα (TOP2A), upregulating growth arrest and DNA damage-inducible protein 45A (GADD45A), and regulating DNA repair and ribosome formation-related mechanisms. Therefore, these molecular mechanisms contribute to the development of Dox resistance in osteosarcoma. Furthermore, in pancreatic adenocarcinoma (PAAD), U3 snoRNA-associated proteins, rRNA processing protein 9 (RRP9)/U3-55K induce gemcitabine resistance. This protein activates the AKT signaling pathway, reducing DNA damage and hindering apoptosis. Consequently, the combination of the AKT inhibitor MK-2206 with gemcitabine shows potential as a therapeutic strategy for gemcitabine-resistant pancreatic cancer patients.
Cell death pathways, such as apoptosis, autophagy, and necrosis, exhibit distinct morphological and biochemical characteristics. Apoptosis is typically associated with cell shrinkage, while autophagy is characterized by cytoplasmic vacuolization and the formation of autophagosomes. These autophagosomes facilitate the elimination of substances through lysosomes. SnoRNAs may influence tumor progression by regulating cell death. SNORD88C promotes the growth and metastasis of non-small cell lung cancer (NSCLC) by inducing 2′-O-methylation of 28S rRNA. This process enhances the translational activity of the downstream target gene SCD1, reduces the number of autophagosomes, and inhibits autophagy. Moreover, the disruption of snoRNA-mediated apoptosis mechanisms affects the resistance of tumor cells to anticancer drugs. SNORD33 binds to methyl-CpG binding protein 2 (MeCP2) and disrupts its interaction with target genes. Consequently, the expression of anti-apoptotic proteins (myeloid leukemia 1 (MCL-1) and BCL-2) decreases, while the expression of pro-apoptotic proteins (caspase-3 (Cas3) and Cas9) increases. Therefore, promoting tumor cell apoptosis leads to the reversal of cisplatin resistance in triple-negative breast cancer (BRCA) cells.
ROS (reactive oxygen species) are produced during oxidative stress processes. Increased cellular ROS levels activate various antioxidant mechanisms in tumor cells. This suppresses ROS production, thereby increasing resistance to cancer treatment. Previous studies have demonstrated the role of snoRNAs in regulating cellular oxidative stress and promoting tumor resistance. The orphan snoRNA ACA11 inhibits oxidative stress, promotes the proliferation of multiple myeloma (MM) cells, and enhances resistance to Dox and 5-FU. ACA11 inhibits the transcriptional activity of the nuclear factor erythroid 2-related factor 2 (NRF2) in multiple myeloma. This inhibition increases ROS-dependent biological processes, including the synthesis of precursor 47S rRNA, protein synthesis, and ribosome generation. Furthermore, the overexpression of ACA11 enhances the responsiveness of MM cells to proteasome inhibitors such as bortezomib.
Small nucleolar RNAs (snoRNAs) can regulate the expression of multidrug resistance-associated proteins (MRP1)/ATP-binding cassette sub-family C member 1 (ABCC1), thereby affecting intracellular drug transport and promoting multidrug resistance in cancer cells. In pancreatic cancer, hsa-miR-1291 derived from SNORA34 specifically targets the 3′ untranslated region (3′ UTR) of the membrane transport protein ABCC1, leading to its downregulation. Consequently, intracellular drug accumulation increases, resulting in the reversal of cancer cell resistance to mitoxantrone.
(Image source: Journal of Pharmaceutical Analysis)
(Image source: Journal of Pharmaceutical Analysis)
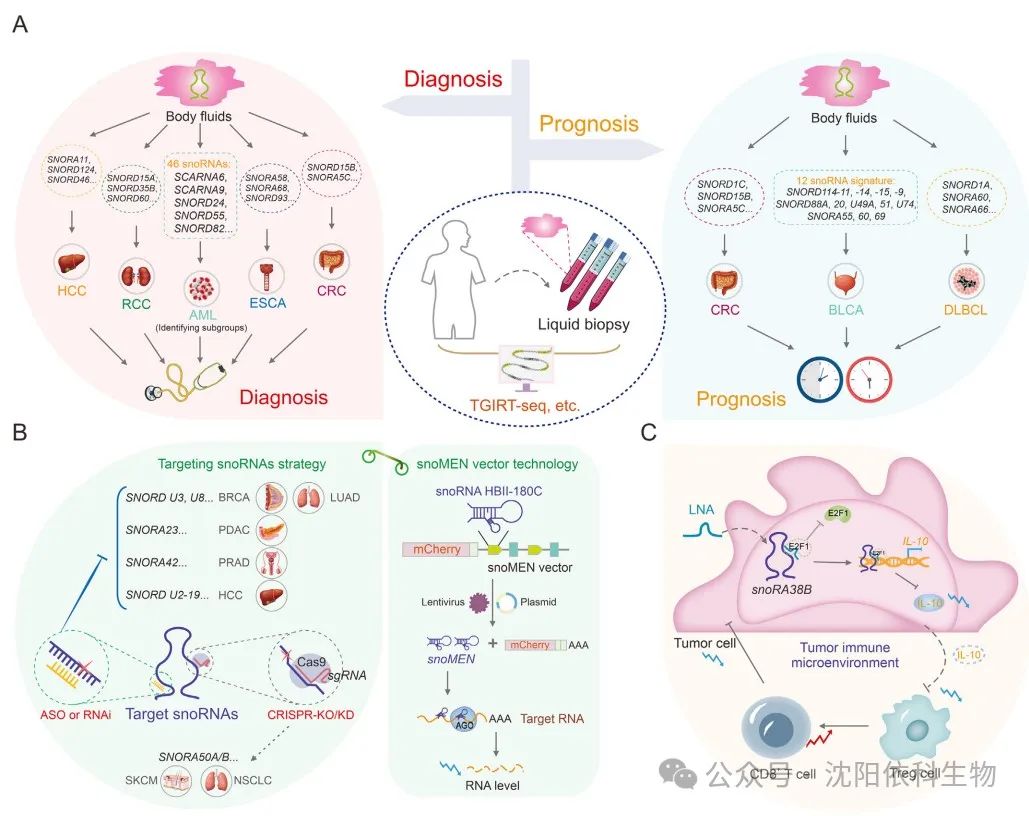
SnoRNAs play significant roles in cancer and can be easily detected in various body fluids, including plasma and serum. With the rapid development of high-throughput sequencing and bioinformatics technologies, snoRNAs have become promising diagnostic and prognostic biomarkers in recent years. Notably, the heat-resistant group II intron reverse transcription RNA sequencing (TGIRT-seq) has high fidelity, sustained synthesis capability, and strand displacement activity, as well as skilled template conversion capabilities. Evidence suggests that TGIRT-seq can comprehensively analyze structured small non-coding RNAs (sncRNAs), including snoRNAs, fundamentally changing the understanding of snoRNA expression. This method provides a new strategy for assessing tumor occurrence and outcomes. Moreover, by analyzing data from The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) datasets, SNORA11, SNORD124, and SNORD46 have been identified as key snoRNAs with potential significance in the diagnosis and treatment of hepatocellular carcinoma (HCC). Similarly, SNORD15A, SNORD35B, and SNORD60 are significantly overexpressed in cancer tissues and urine, making them useful biomarkers for early diagnosis and potential therapeutic targets in renal cell carcinoma (RCC). In esophageal cancer (ESCA), SNORA58, SNORA68, and SNORD93 have been observed to be upregulated in ESCA patients and early patients. Furthermore, the combination of snoRNAs with existing tumor markers, such as carcinoembryonic antigen, can enhance the diagnostic capability for ESCA patients. In diffuse large B-cell lymphoma (DLBCL), a triple snoRNA prognostic risk model composed of SNORD1A, SNORA60, and SNORA66 has been constructed. The study reveals that the upregulation of SNORD1A and SNORA60 is associated with poor prognosis in DLBCL patients, while SNORA66 is upregulated in the low-risk group, providing a protective effect. This study offers new insights into the prognostic significance of snoRNAs in DLBCL. The oncogenic characteristics of SNORD15B and SNORA5C in colorectal cancer (CRC) suggest that these two snoRNAs could serve as promising biomarkers for the diagnosis and prognostic prediction of CRC. Furthermore, different B-cell malignancies exhibit abnormal expression of specific snoRNAs. Using large-scale parallel sequencing (through oligonucleotide ligation and detection sequencing (SOLiD)), 46 snoRNAs that are differentially expressed between precursor B-cell acute lymphoblastic leukemia (BCP-ALL) and T-cell acute lymphoblastic leukemia (T-ALL) have been identified. These snoRNAs have potential applications as diagnostic biomarkers or therapeutic targets. SnoRNAs exhibit various properties in cancer, serving as reliable prognostic markers. In bladder cancer (BLCA), a risk assessment classifier based on the expression profile of 12 snoRNAs associated with survival has been developed. Clinical outcomes differ between high-risk and low-risk patient groups. This characteristic makes it a reliable prognostic predictor for BLCA patients. By analyzing the comprehensive expression profiles of snoRNAs across 21 purified immune cell lines, 43 colorectal cancer cell lines, and three datasets, snoRNA features associated with immune infiltration (TIIsno-snoRNA) have been identified. Patients with lower TIIsno scores respond better to immunotherapy. This feature may serve as an indicator for predicting the prognosis of colorectal cancer patients and their response to immunotherapy.
SnoRNAs participate in the carcinogenic process through various regulatory mechanisms. Due to their oncogenic potential, these molecules may serve as targets for cancer therapy. Various cancer treatment strategies targeting snoRNAs have been developed, including clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, small interfering RNA (siRNA), short hairpin RNA (shRNA), and antisense oligonucleotide (ASO) technologies. ASOs can effectively silence non-coding RNAs. ASOs may serve as effective and targeted therapeutic interventions, specifically targeting cancer-related snoRNAs. Additionally, ASOs can suppress the tumorigenicity of cancer cells in vitro and in vivo. In vitro experiments indicate that targeting box C/D snoRNAs U3 and U8 can eliminate the tumorigenicity of lung (H1944) and breast cancer (MCF-7) cells. SNORA23 upregulates SYNE2 expression, promoting the survival and invasiveness of pancreatic ductal adenocarcinoma (PDAC) cells. The use of ASOs targeting SNORA23 can reduce the growth and metastasis of xenograft tumors. Therefore, ASO technology holds promise as a novel cancer treatment method targeting tumor-associated snoRNAs. Both siRNAs and shRNAs can induce target gene degradation and inhibit tumor progression by degrading oncogenes. Indeed, siRNAs and shRNAs are relatively inefficient against highly structured mRNAs, but due to the conserved secondary structures and crucial RNA-binding sequences of snoRNAs, they can effectively silence snoRNAs. For example, SNORA42 is significantly overexpressed in prostate cancer tissues compared to adjacent tissues. Transfecting anti-SNORA42 siRNA significantly reduces the growth rate of DU145 and PC3 prostate cancer cells and induces apoptosis. The increased expression of snoU2_19 is associated with an aggressive phenotype in HCC patients. Furthermore, using snoU2_19-shRNA to knock down snoU2_19 effectively inhibits the cell cycle proliferation and progression of SK-HEP-1 and HCCLM3 HCC cells. Additionally, CRISPR/Cas9 gene editing technology has been used to remove snoRNAs, particularly SNORA50 A/B, leading to increased tumorigenicity in CHL1 melanoma, A549 lung cancer, and NCI-H23 cells. Furthermore, snoRNA gene expression regulators (snoRNA gene expression regulatory modules (snoMEN)), a snoRNA-derived vector technology, may also be beneficial for targeted cancer therapy. The snoMEN technology, as an antisense gene suppression technique, targets the intronic sequences of precursor mRNAs, leading to downregulation of genes and regulation of RNA targets. The snoMEN vector selectively induces the overexpression of miR21 in human lung adenocarcinoma cells by targeting specific sequences in pri-miR21, inducing apoptosis.
SnoRNAs are associated with multiple components of the tumor immune microenvironment, including CD8+ T-cell infiltration, cytotoxic T-cell function, and tumor angiogenesis. Targeting snoRNAs may enhance the efficacy of cancer immunotherapy. SNORA38B promotes the recruitment of CD3+FOXP3+ regulatory T cells (Tregs) into the tumor microenvironment of non-small cell lung cancer (NSCLC) by stimulating the secretion of interleukin-10 (IL-10). This results in reduced infiltration of CD3+CD8+ T cells, thereby promoting tumor development and immune suppression. Targeting SNORA38B with locked nucleic acids (LNAs) can significantly improve the tumor immune microenvironment and enhance the responsiveness of non-small cell lung cancer cells to immune checkpoint blockade therapy.
(Image source: Journal of Pharmaceutical Analysis)
SnoRNAs were once overlooked and only considered a subtype of non-coding RNAs (ncRNAs) in cancer cells. However, in recent years, there has been increasing interest in snoRNAs and their roles in tumor development, particularly their functions in 2′-O-ribose methylation and pseudouridylation. Nevertheless, a comprehensive understanding of the complex relationship between snoRNAs and cancer remains limited. Multiple studies indicate that snoRNAs exhibit atypical characteristics and expression patterns. This review introduces the regulatory mechanisms of snoRNA expression from three perspectives: the dynamic regulation by transcriptional regulators, the repression of transcription by DNA methylation, and the dependence on host gene transcription and splicing events. Importantly, it briefly outlines and explains three novel molecular mechanisms regulating cancer-related snoRNAs: 1) the transcriptional regulation of targets by snoRNAs, including interactions between snoRNAs and transcriptional regulators (TRs); 2) the regulatory roles of snoRNAs in post-transcriptional processes, including precursor RNA splicing, mRNA 3′ end APA, mRNA stability, and the formation of sdRNAs with miRNA-like functions, which indirectly regulate tumor occurrence and development; 3) the effects of snoRNAs on the post-translational ADP-ribosylation of target RNAs and chromatin remodeling. The non-classical cancer regulatory mechanisms of snoRNAs undoubtedly enhance the understanding of cancer and open new avenues for tumor therapy. SnoRNAs may influence the drug resistance of tumor cells by regulating stemness, DNA damage repair (DDR), cell death pathways, and oxidative stress. Therefore, they can be viewed as promising therapeutic targets in cancer treatment, particularly those associated with drug resistance. Moreover, the interactions and feedback mechanisms between SNHG (snoRNA host genes) and snoRNAs have significant impacts on cancer progression, warranting further investigation.
(Image source: Journal of Pharmaceutical Analysis)
(Image source: Journal of Pharmaceutical Analysis)