AI-Empowered Materials

In recent years, the application of Metal-Organic Frameworks (MOFs) in the field of adsorption has transitioned from proof-of-concept research to practical technological applications. Some MOFs, such as CALF-20 and MOF-303, have achieved kilogram-scale usage, respectively for CO2 capture from flue gas and atmospheric water extraction from desert air. Among them, MOF-303, as a highly promising water collection material, exhibits an S-shaped adsorption curve at low vapor pressures, which arises from the strong interactions between adjacent PZDC ligand pyrazole functional groups acting as nucleation sites. However, this low-pressure adsorption reduces its working capacity. By replacing the PZDC ligand with a less hydrophilic FDC ligand to obtain MOF-333, this issue is resolved, as its adsorption isotherm shows a steep step, with the relative humidity at which the step occurs (22%) being suitable for arid desert environments. Meanwhile, MOF-LA2−1 enhances the working capacity by approximately 50% compared to MOF-303 through a PZDC ligand extension strategy. To rationally optimize the adsorption performance of MOFs and guide their modification for target applications, a deep understanding of the intermolecular interactions and regulatory mechanisms affecting adsorption behavior is essential. Therefore, computational studies predicting adsorption isotherms are crucial. Currently, the commonly used methods for predicting adsorption isotherms are Grand Canonical Monte Carlo (GCMC) or Gibbs Ensemble Monte Carlo (GEMC) simulations, but they often rely on classical force fields, which suffer from insufficient potential energy surface descriptions and inadequate consideration of framework flexibility, making it difficult to meet precise prediction requirements. To address this, this study proposes an integrated workflow that combines high-accuracy and data-efficient Machine Learning Potentials (MLP) with Transition Matrix Monte Carlo (TMMC) simulations to achieve chemically accurate modeling of the water adsorption process in flexible MOFs, providing a powerful tool for the network design of MOFs.
Article Summary

Metal-Organic Frameworks (MOFs) such as MOF-303 and MOF-LA2−1 exhibit excellent performance in water collection applications. To achieve the network design of such materials, it is necessary to accurately predict their adsorption performance with chemical precision while fully considering framework flexibility. Currently, the computational prediction of water adsorption performance in MOFs has become a routine method, but existing approaches lack the predictive capability required for designing new materials due to insufficient descriptions of interatomic potential energy and inadequate consideration of framework flexibility. This study proposes a method to obtain chemically accurate adsorption isotherms while fully considering framework flexibility, relying on high-precision and efficiently trained Machine Learning Potentials (MLP) and Transition Matrix Monte Carlo (TMMC) simulations. For MOF-303, if the MLP is trained using accurately benchmarked electronic structure methods and considers local and global framework flexibility, quantitatively accurate adsorption isotherms can be obtained. The broad applicability of this method is validated through studies on MOF-333 and MOF-LA2−1. Analysis of the water density distribution in MOFs reveals the controlling factors of the isotherm shape and origin: ideal water collection materials should possess nucleation sites with moderate adsorption strength to avoid adverse low-pressure water uptake; while maintaining the characteristics of the initial nucleation sites, the working capacity can be increased through ligand extension strategies (e.g., MOF-LA2−1). This method can also be applied to other guest molecules and MOFs, providing possibilities for designing MOFs with specific adsorption properties.
Author Introduction
This research was jointly completed by Ruben Goeminne and Veronique Van Speybroeck from the Molecular Modeling Center at Ghent University, Belgium, with Veronique Van Speybroeck serving as the corresponding author. Ruben Goeminne’s research focuses on computational simulations of MOF adsorption performance and machine learning modeling, making significant contributions in constructing high-precision MLP models and conducting TMMC simulations to analyze the impact of MOF framework flexibility on adsorption behavior. Veronique Van Speybroeck, as a senior researcher, has long been dedicated to research in molecular modeling and computational chemistry, playing a key role in guiding research directions, designing experimental plans, and ensuring research quality. The two have closely collaborated, combining their respective professional strengths to successfully develop a method for predicting MOF adsorption performance that balances chemical precision and broad applicability, providing important theoretical support for the design and application of MOF materials.
Article Approach
Subsequently, the research direction centered on combining Machine Learning Potentials (MLP) with Transition Matrix Monte Carlo (TMMC) simulations was determined. To establish a reliable computational model, theoretical level benchmark tests were first conducted, selecting various GGA, meta-GGA, and hybrid functionals. By calculating the interaction energies of eight water hexamer clusters and a single water molecule in the adsorption pocket of MOF-303, and comparing them with CCSD(T) reference results, rPBE-D3 (BJ) was selected as the optimal theoretical level, as it exhibited minimal errors in predicting MOF-water and water-water interaction energies, meeting chemical precision requirements. Next, an active learning strategy was employed to train the MLP, constructing initial datasets for MOF-303, MOF-333, and MOF-LA2−1. Through multiple rounds of MLP NPT-MD simulations at different temperatures (100K, 300K, 500K), snapshots were extracted and recalculated using DFT to supplement the training set, ultimately obtaining an MLP model that accurately describes the framework flexibility of MOFs and intermolecular interactions. Subsequently, TMMC simulations were conducted based on the trained MLP, performing MD simulations in the NPT ensemble to fully consider framework flexibility, calculating acceptance probabilities through virtual guest molecule insertion/deletion, and thus obtaining adsorption isotherms. The adsorption performance of MOF-303, MOF-333, and MOF-LA2−1 was simulated and predicted, comparing the effects of different theoretical levels and framework flexibility handling methods on the simulation results, and validating the model accuracy with experimental data. Finally, by analyzing the number of water-framework hydrogen bonds, water-water hydrogen bonds, and water density distribution in the three MOFs, the atomic-level reasons for the differences in isotherm shapes were revealed, extracting design principles for MOF water collection materials, providing guidance for the rational design of subsequent MOFs.
Main Research Content
1. Theoretical Level Screening and Machine Learning Potential (MLP) Training:
To achieve chemically accurate predictions of MOF water adsorption performance, the research team first conducted systematic theoretical level benchmark testing and high-precision MLP model training. Considering the sensitivity of adsorption performance calculation results to the description of intermolecular interactions, the study selected various representative Density Functional Theory (DFT) functionals, including GGA, meta-GGA, and hybrid functionals, to evaluate their performance by calculating two key interaction energies: one is the water-water interaction energy of eight water hexamer clusters, and the other is the MOF-water interaction energy of a single water molecule in the main adsorption pocket of MOF-303, comparing these results with high-precision CCSD(T) reference values. The results showed significant differences in interaction energy predictions among different functionals, with some functionals exhibiting notable underbinding or overbinding issues for both framework-water and water-water interactions, while the rPBE-D3 (BJ) functional performed the best, with an error of only 2.4 kJ/mol for the MOF-water interaction energy (the corresponding MOF-water interaction energy being -103.52 kJ/mol) and an average error of -0.87 kJ/mol for the water-water interaction energy, thus meeting chemical precision requirements and being determined as the reference theoretical level for subsequent MLP training.
During the MLP training process, the research employed an efficient active learning strategy to avoid the time-consuming issues associated with traditional DFT MD simulations. For MOF-303, an initial dataset was constructed by inserting 0-42 water molecules into a 2×2×2 supercell empty framework (ensuring that interatomic distances met van der Waals radius requirements), repeating this process 10 times to obtain 430 structures, applying two random position and cell perturbations to each structure, ultimately generating 860 snapshots for initial MLP training. Subsequently, three phases of active learning cycles were conducted: the first phase involved 5 rounds of MLP NPT-MD simulations at 100K (1000 steps per round, step size 0.5fs), where low-temperature conditions prevented model instability due to sampling exceeding the training set phase space; the second phase raised the temperature to 300K, conducting 5 rounds of simulations (4000 steps per round); the third phase further increased the temperature to 500K, conducting 5 rounds of simulations (10000 steps per round). At the end of each simulation round, the final MD snapshots were extracted, and energies and forces were recalculated at the rPBE-D3 (BJ) level using VASP software, adding them to the training set to update the MLP model. For MOF-333 and MOF-LA2−1, the initial dataset construction method was adjusted based on their structural characteristics: MOF-333 considered both cis and trans ligand conformations, with each adsorption amount corresponding to 5 snapshots containing guest molecules; MOF-LA2−1 encompassed 16 possible ligand conformations, selecting 26 intermediate adsorption amounts between 0-68 for each conformation, generating 5 snapshots for each adsorption amount. The final trained MACE MLP model maintained validation errors for energy and force within 0.9 meV/atom and 43 meV/Å, respectively, accurately describing the flexible characteristics of MOF frameworks and intermolecular interactions.
2. Transition Matrix Monte Carlo (TMMC) Simulation and Adsorption Performance Validation:
Based on the trained high-precision MLP model, the research team conducted MOFs water adsorption performance predictions through TMMC simulations, validating the accuracy and reliability of the model with experimental data. TMMC simulations consist of three key steps: first, for each adsorption amount, 20 snapshots of MOFs containing guest molecules were selected (10 for MOF-333 and MOF-LA2−1), and balanced Monte Carlo simulations were performed using MLP in the NVT ensemble, executing 5000 MC moves for each adsorbate, including molecular reinsertion, local translation, and rotation, to ensure the system reached a balanced state; next, for each balanced snapshot, OpenMM software was used to conduct 150 ps of NPT-MD simulations at 298K and 0MPa (step size 0.833fs), fully considering the local and global flexibility of the MOF framework and capturing structural changes during the adsorption process; finally, snapshots were extracted from each MD simulation trajectory, attempting 10⁶ virtual guest molecule insertions and deletions to calculate their acceptance probabilities, thereby constructing a collection matrix and obtaining macrostates probabilities for different adsorption amounts at various gas pressures, ultimately generating adsorption isotherms.
To comprehensively validate the model performance, the study first used MOF-303 as a benchmark, comparing the effects of different theoretical levels (PBE-D3 (BJ), revPBE-D3 (BJ), rPBE-D3 (BJ), etc.) and different framework flexibility handling methods (fully flexible, only local flexibility, no flexibility) on the simulation results. The results indicated that simulations based solely on the rPBE-D3 (BJ) theoretical level and considering full framework flexibility could accurately predict the shape of the MOF-303 adsorption isotherm and the initial pressure of the adsorption step, quantitatively matching experimental results; other theoretical levels exhibited deviations in adsorption amount predictions due to overbinding or underbinding issues, while neglecting framework flexibility caused the S-shaped characteristics of the isotherm to disappear or significantly delayed the adsorption step. Subsequently, this method was applied to MOF-333 and MOF-LA2−1: for MOF-333, the simulation results of the cis ligand conformation at the rPBE-D3 (BJ) level were closest to the experimental isotherm, successfully reproducing its steep adsorption step; for MOF-LA2−1, the simulation results of the ZUS series ligand conformations overall outperformed those of the ENT series, and all conformations reasonably reflected its higher saturated adsorption amount, slightly exceeding the experimental value due to the assumption of perfect crystallization in the simulation. Through multi-system and multi-condition validation, the method’s chemical precision and broad applicability in predicting the water adsorption performance of flexible MOFs were fully demonstrated.
3. Adsorption Mechanism Analysis and Extraction of MOFs Design Principles:
To reveal the essence of the differences in MOFs adsorption behavior at the atomic level and guide the design of high-performance water collection MOFs, the research team conducted in-depth analyses of hydrogen bonding interactions, water density distributions, and nucleation behaviors in three MOFs (MOF-303, MOF-333, MOF-LA2−1), extracting key design principles. In the analysis of hydrogen bonding interactions, by statistically examining the number of MOF-water hydrogen bonds and water-water hydrogen bonds as a function of adsorption amount, it was found that at low adsorption amounts, both MOF-303 and MOF-LA2−1, due to the presence of pyrazole functional groups in their ligands, could form approximately 4 strong MOF-water hydrogen bonds per adsorbate, leading to significant water uptake at low pressures; whereas MOF-333, due to the use of the less hydrophilic FDC ligand, exhibited weaker MOF-water hydrogen bonding, resulting in minimal water uptake at low pressures. At high adsorption amounts, the rate of decrease of MOF-water hydrogen bonds in MOF-LA2−1 was faster than that in MOF-303, becoming closer to MOF-333, which is consistent with its higher initial pressure for the adsorption step (approximately 26% RH); the trends in the number of water-water hydrogen bonds for the three MOFs were similar, indicating a relatively uniform influence of water-water interactions on adsorption behavior.
The three-dimensional visualization and statistical analysis of water density distributions further revealed the correlation between nucleation site characteristics and adsorption isotherm shapes: at low adsorption amounts, both MOF-303 and MOF-LA2−1 formed four high-density nucleation regions near the pyrazole functional groups, corresponding to strong adsorption sites, which is the root cause of their S-shaped isotherms; MOF-333, on the other hand, exhibited eight low-density nucleation regions, doubling the number of nucleation sites but reducing adsorption strength, effectively avoiding low-pressure water uptake and forming a steep adsorption step. As the adsorption amount increased, the density of nucleation sites in MOF-303 and MOF-LA2−1 decreased due to the formation of a water-water hydrogen bond network, while the nucleation site density in MOF-333 increased, further enhancing its steep adsorption characteristics. Based on the above analysis, the core design principles for water collection MOFs were distilled: first, the initial nucleation sites should possess moderate adsorption strength to ensure efficient water uptake at target relative humidity while avoiding adverse low-pressure water uptake; second, while maintaining the characteristics of the initial nucleation sites, the working capacity can be increased through ligand extension strategies (e.g., MOF-LA2−1 extending the PZDC ligand to PZVDC) to enlarge pore volume and enhance saturated adsorption amounts and working capacity. These design principles provide clear theoretical guidance for developing the next generation of high-performance water collection MOFs through network synthesis strategies.
Illustrated Guide
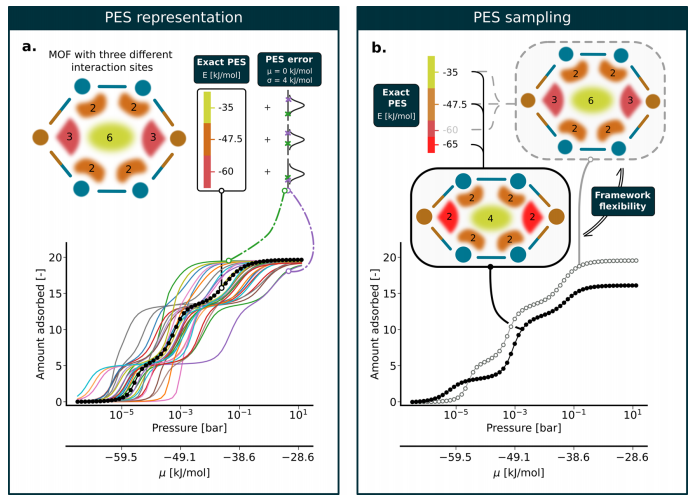
Figure 1: (a) Shows a MOF toy system with three different interaction sites, with a color bar indicating the corresponding interaction energies, each site labeled with the maximum number of adsorbates, and the black dashed line representing the accurately calculated adsorption isotherm at 273K, while the colored curves indicate the isotherms after adding a 4 kJ/mol width normal distribution error to each interaction site, demonstrating that even within the range of chemical precision, potential energy errors can lead to significant deviations in isotherm shapes and adsorption jump pressures; (b) Displays the impact of interaction energy changes caused by framework flexibility on adsorption isotherms, where narrowing pores increases the interaction strength at the left and right nodes but reduces the maximum adsorption amount from 3 to 2, and the maximum adsorption amount at the pore center decreases from 6 to 4, ultimately leading to a one-order-of-magnitude reduction in the initial adsorption pressure of the isotherm and the appearance of a plateau before 10⁻³ bar, reflecting the significant impact of framework flexibility on adsorption behavior.
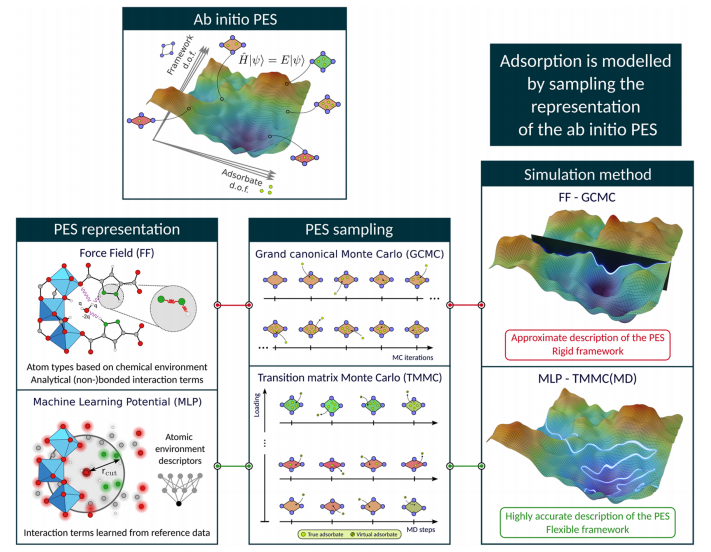
Figure 2: This figure illustrates the two core issues that need to be addressed in modeling the adsorption process: the “PES Representation” module on the left compares the differences between traditional force fields (FF) and MLP: FF approximates PES descriptions based on atomic types and analytical bonding/non-bonding terms, assuming framework rigidity; MLP learns interaction terms from reference data through atomic environment descriptors, enabling high-precision descriptions of the free energy surface (FES) and handling flexible frameworks. The “PES Sampling” module in the middle compares GCMC and TMMC: GCMC requires gradual addition/deletion of molecules, which is easily limited by free energy barriers; TMMC efficiently samples by conducting simulations for each adsorption amount in the NVT ensemble without the need for gradual addition of molecules. The red line connects the commonly used FF-GCMC method in the literature, while the proposed MLP-TMMC method (combining MLP’s high-precision PES description with TMMC’s efficient flexible sampling) achieves chemically accurate adsorption simulations.
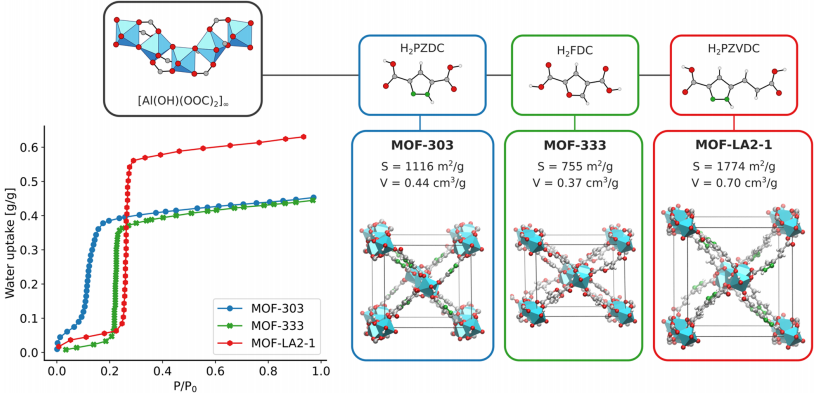
Figure 3: Displays the experimental adsorption isotherms of MOF-303, MOF-333, and MOF-LA2−1, while also showcasing the organic ligands of the three MOFs, the DFT-optimized atomic structures, and the specific surface area (S) and pore volume (V) calculated using PoreBlazer: MOF-303 has a ligand of PZDC, S=1116 m²/g, V=0.44 cm³/g; MOF-333 has a ligand of FDC, S=755 m²/g, V=0.37 cm³/g; MOF-LA2−1 has a ligand of PZVDC, S=1774 m²/g, V=0.70 cm³/g. From the isotherms, it can be seen that MOF-303 exhibits S-shaped adsorption at low pressures, MOF-333 shows a steep step in the adsorption isotherm, and MOF-LA2−1 has a higher saturated adsorption amount, with the differences in adsorption behavior among the three closely related to the ligand structure and pore structure characteristics.
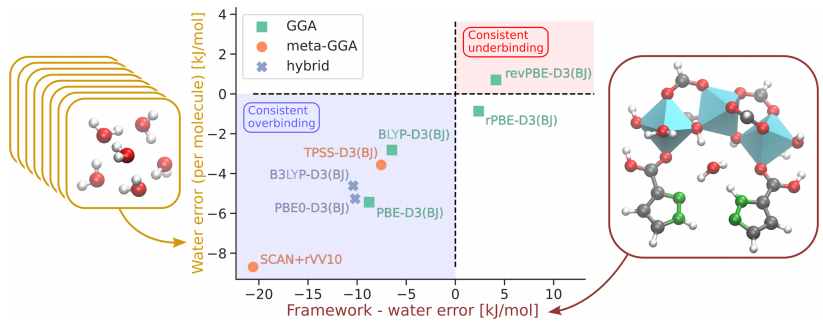
Figure 4: This figure compares the performance of different DFT functionals relative to CCSD(T) calculation results, with the Y-axis representing the error in water-water interaction energy (based on calculations of eight water hexamers) and the X-axis representing the error in framework-water interaction energy (based on calculations of a single water molecule in the main adsorption site of MOF-303). The figure displays the error distribution of the selected GGA, meta-GGA, and hybrid functionals, with functionals in the upper right region exhibiting underbinding issues for both framework-water and water-water interactions, while those in the lower left region exhibit overbinding issues. Among them, the rPBE-D3 (BJ) functional performed the best, with an error of 2.4 kJ/mol for the framework-water interaction energy (the framework-water interaction energy being -103.52 kJ/mol) and an average error of -0.87 kJ/mol for the water-water interaction energy, providing a reliable theoretical level choice for subsequent MLP training.
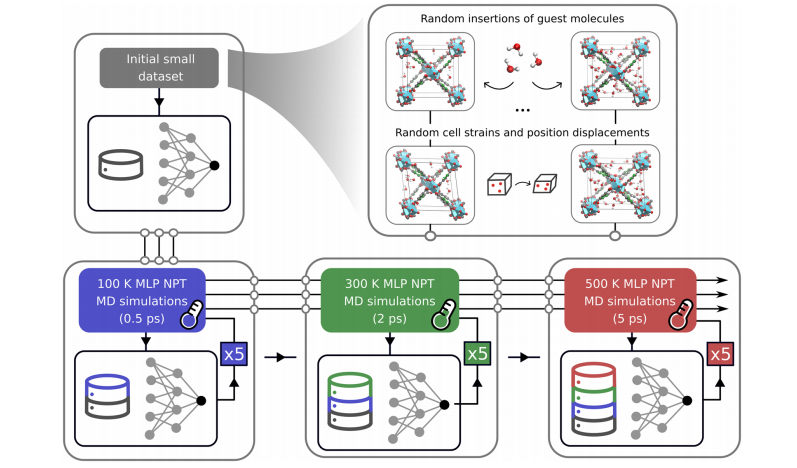
Figure 5: Displays the active learning cycle process employed. The initial dataset consists of MOF snapshots with added guest molecules, to which random cell strains and position displacements are applied; subsequently, three groups of active learning iterations are conducted: first, 5 rounds of MLP NPT-MD simulations (each round 0.5 ps) at 100K, followed by 5 rounds at 300K (each round 2 ps), and finally 5 rounds at 500K (each round 5 ps). After each iteration, the final frame of the MD simulation is extracted, recalculated at the reference DFT level, and added to the training set, gradually increasing the simulation temperature to expand the phase space sampling range, ultimately obtaining an MLP model that accurately describes the flexibility of MOF frameworks and intermolecular interactions.
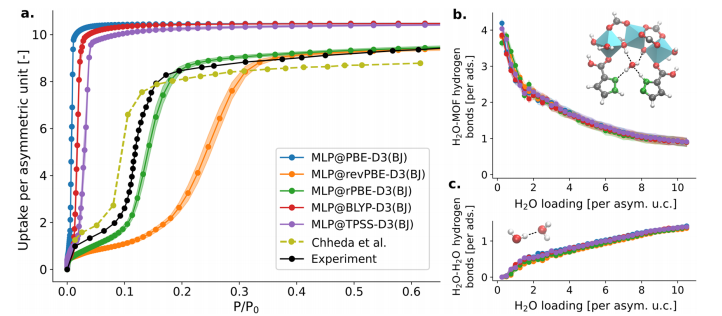
Figure 6: (a) Shows the adsorption isotherm of MOF-303 obtained through TMMC simulations using MLP trained at five selected theoretical levels at 298K, comparing the experimental results with Gibbs ensemble Monte Carlo simulation results based on force fields. Only the rPBE-D3 (BJ) level accurately predicts the initial pressure of the adsorption step, while other theoretical levels exhibit over-prediction or under-prediction issues; (b) and (c) respectively show the number of MOF-water hydrogen bonds and water-water hydrogen bonds calculated at each theoretical level as a function of water adsorption amount. The trends in hydrogen bond numbers predicted by all theoretical levels are similar, with approximately 4 MOF-water hydrogen bonds per adsorbate at low adsorption amounts, decreasing to about 0.9 per adsorbate at high adsorption amounts, while water-water hydrogen bonds are approximately 1.4 per adsorbate at high adsorption amounts, indicating that even with energy errors at the theoretical level, a qualitatively correct understanding of interaction sites and water structure can still be obtained.
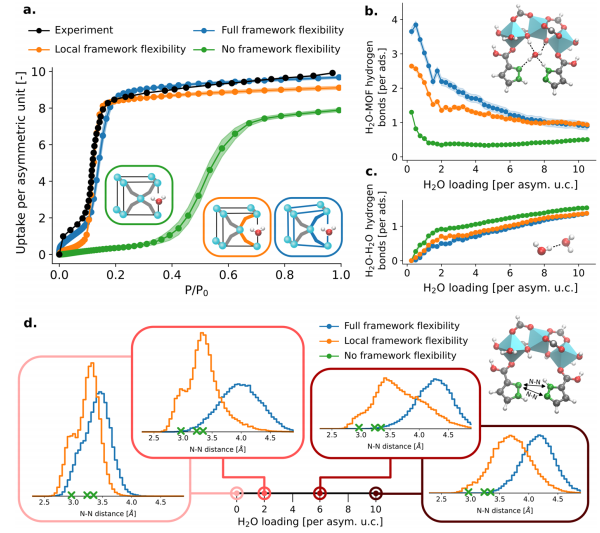
Figure 7: (a) Shows the adsorption isotherm of MOF-303 at 298K based on rPBE-D3 (BJ) MLP-TMMC simulations, calculated using constrained host NVT (green, no framework flexibility), NVT (orange, only local flexibility), and NPT (blue, full flexibility) trajectories. The full flexibility simulation results best match the experimental data, while the isotherm’s S-shaped characteristics disappear or the adsorption step is delayed when only local flexibility or no flexibility is considered; (b) and (c) respectively show the changes in the number of MOF-water hydrogen bonds and water-water hydrogen bonds as a function of adsorption amount under different flexibility handling methods. Under full flexibility treatment, the number of MOF-water hydrogen bonds at low adsorption amounts is approximately 4 per adsorbate, dropping below 1.5 when no flexibility is considered, while the number of water-water hydrogen bonds slightly increases; (d) Shows the radial distribution function of nitrogen-nitrogen distances as a function of adsorption amount under different flexibility handling methods, remaining constant (green cross marks) when no flexibility is considered, while under full flexibility, the nitrogen-nitrogen distance increases with increasing adsorption amount, and is underestimated under only local flexibility, indicating that framework flexibility is crucial for “opening” the adsorption pocket and adsorption behavior.
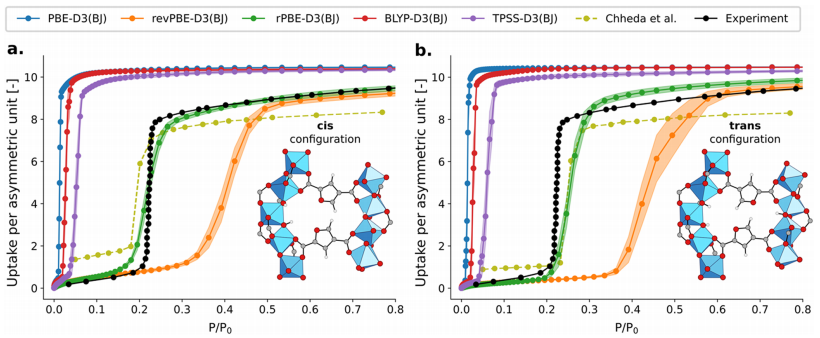
Figure 8: Shows the adsorption isotherms of MOF-333 cis (a) and trans (b) ligand conformations at 298K obtained through TMMC simulations based on MLP trained at five selected theoretical levels, comparing the experimental results with Gibbs ensemble Monte Carlo simulation results based on force fields. Similar to MOF-303, the simulation results at the rPBE-D3 (BJ) theoretical level best match the experimental data, with the cis ligand conformation’s simulation results being closer to the experimental data, confirming the robustness of the method across different ligand conformation MOFs, while also highlighting the importance of using flexible models and precise MLP to improve isotherm prediction accuracy.
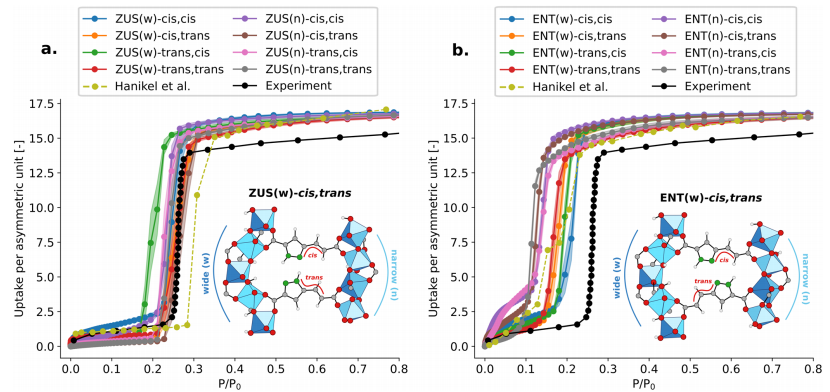
Figure 9: Shows the adsorption isotherms of MOF-LA2−1 ZUS (a) and ENT (b) ligand conformations at 298K obtained through TMMC simulations based on rPBE-D3 (BJ), comparing the experimental results with Gibbs ensemble Monte Carlo simulation results based on force fields. There are subtle differences in the adsorption isotherms of different ligand conformations, but the ZUS conformation is overall closer to the experimental results, consistent with the conclusions of Hanikel et al.; all ligand conformations exhibit overestimation of saturated adsorption amounts, which is speculated to be related to the lower crystallinity of MOF-LA2−1 in experiments, while the assumption of perfect crystallization in simulations further validates the reliability of the method and the impact of experimental conditions on simulation results.
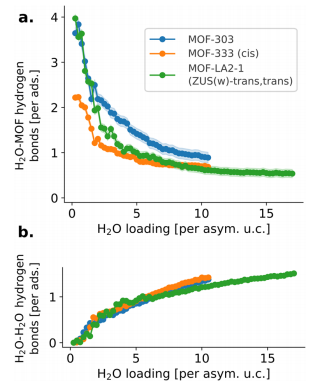
Figure 10: Shows the changes in the number of MOF-water hydrogen bonds (a) and water-water hydrogen bonds (b) as a function of water adsorption amount for MOF-303 (blue), MOF-333 cis conformation (orange), and MOF-LA2−1 ZUS (w)-trans conformation (green). At low adsorption amounts, both MOF-303 and MOF-LA2−1, due to the presence of pyrazole groups in their ligands, have approximately 4 MOF-water hydrogen bonds per adsorbate; at high adsorption amounts, the MOF-water hydrogen bonds in MOF-LA2−1 decrease more rapidly, becoming closer to MOF-333, which also leads to its relatively higher pressure for the adsorption step (approximately 26%); the trends in the number of water-water hydrogen bonds for the three MOFs are similar, indicating a relatively consistent influence of water-water interactions on adsorption behavior.
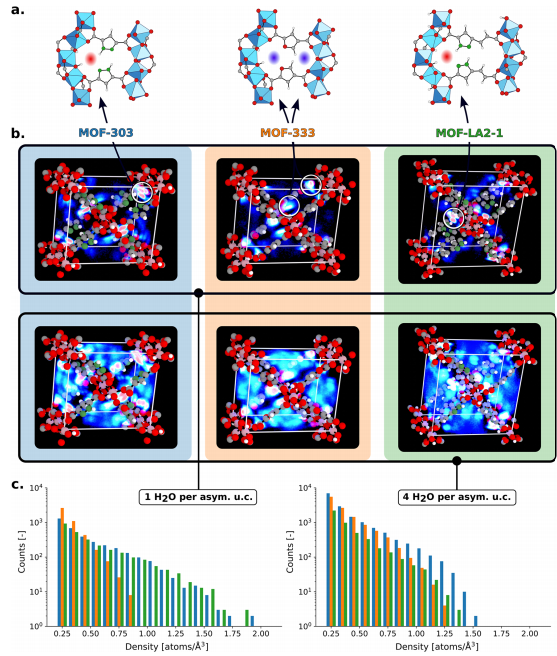
Figure 11: (a) Illustrates the distribution differences of nucleation sites between the organic ligands of the three MOFs; (b) Shows the three-dimensional density distribution of water in MOF-303, MOF-333 cis conformation, and MOF-LA2−1 ZUS (w)-trans conformation when containing 1 (upper row) or 4 (middle row) adsorbates in each asymmetric unit, with blue and red representing medium and high adsorbate densities, respectively. Both MOF-303 and MOF-LA2−1 have four high-density regions (red) when containing one adsorbate, corresponding to strong nucleation sites near the pyrazole functional groups, while MOF-333 has eight low-density regions (light blue); (c) Shows the water density histograms for the three MOFs when containing 1 (left) or 4 (right) adsorbates in each asymmetric unit, with the density distributions of MOF-303 and MOF-LA2−1 being similar, while the maximum density of MOF-333 is about half that of the former two, with the nucleation site density in MOF-333 increasing at high adsorption amounts, while that in MOF-303 and MOF-LA2−1 decreases due to the effects of water-water hydrogen bonding.
Content Outlook
The method proposed in this study, combining Machine Learning Potentials (MLP) with Transition Matrix Monte Carlo (TMMC) simulations, provides an effective approach for chemically accurate predictions of the adsorption performance of flexible MOFs, with potential for further expansion in multiple aspects. In terms of model optimization, the scale and diversity of the training dataset can be increased, incorporating more data on MOFs with different structures and interactions with various guest molecules, while exploring more advanced MLP architectures and training strategies to further enhance the model’s descriptive accuracy for complex MOF systems (e.g., MOFs with defects or mixed ligands). In terms of application scenario expansion, in addition to water adsorption, this method can be extended to the adsorption simulations of other guest molecules such as CO2 and methane in MOFs, providing support for the design of MOFs in carbon capture, gas storage, and other fields; it can also be combined with experimental synthesis to conduct high-throughput computational screening, accelerating the development process of MOFs with specific functions. In terms of deepening mechanistic research, this method can be utilized to explore the structure-activity relationships between MOF framework flexibility and adsorption performance, revealing the diffusion pathways and adsorption kinetics of guest molecules in MOF channels, providing more precise theoretical guidance for the structural modification and performance tuning of MOF materials. Additionally, attempts can be made to combine this method with other computational chemistry methods (e.g., molecular dynamics simulations, density functional theory) to construct multi-scale computational models, comprehensively analyzing the adsorption mechanisms of MOFs and promoting the widespread application of MOF materials in energy, environment, and other fields.
Note: The content pushed by this public account is for the purpose of communication and learning, not for commercial use. If there is any infringement, please contact us for negotiation. Experts and scholars are welcome to submit articles to share the latest research results in AI + materials!
Submission Email: [email protected]
Group Chat: AI + Materials Research Group
How to Join: Add the editor’s WeChat: FC13296996229, please note: Institution-Name-Research Direction, and the editor will review and invite you to join the group.
Editor’s Note: Spring Doctor Growth Camp 6.4


Public Account丨AI-Empowered Materials
Editor丨Dr. Chun

(Dr. Chun)
Editorial Office Information

READING BOOKS


WeChat ID丨Yao Min
WeChat ID丨FC13296996229