Hello everyone, today I would like to share a recent article published in the Journal of the American Chemical Society, titled:Tuning Entanglement Molecular Weights with Ultrahigh-Molecular Weight Polystyrenics. The corresponding authors of this article are Professors Brent S. Sumerlin and Austin M. Evans from the University of Florida, USA.
The physical entanglement between polymer chains is crucial for determining their thermomechanical properties. When the molecular weight (Mn) exceeds the entanglement molecular weight (Me), an entangled network forms, enhancing high-temperature stability (Figure 1B). Traditional thermoplastics flow easily at high temperatures, while thermosetting materials are stable but cannot be reprocessed. Dynamic covalent crosslinked networks attempt to balance both, but still face issues like creep. This study constructs a reversible physical entanglement network by regulating the ultrahigh molecular weight (UHMW) polymers’ Me, achieving a combination of high thermal stability and reprocessability.
In recent years, breakthroughs in reversible deactivation radical polymerization (especially photopolymerization techniques) have provided new pathways for preparing structurally controllable UHMW polymers. The authors innovatively employed an electron-mismatched alkoxy styrene (electron-rich) and pentafluorostyrene (PFS, electron-deficient) copolymerization strategy, successfully synthesizing UHMW polymers using their rapid cross-propagation characteristics, while the PFS units’ SNAr sites provided conditions for post-modification control of Me (Figure 1B).
The researchers synthesized three different alkyl chain lengths (n=6,12,18) of PASX-PFS ultrahigh molecular weight polymers (Mn=2.9-4.89MDa, dispersity 1.2-1.5, Figure 1B). Rheological tests indicated that these polymers exhibit high entanglement characteristics (Ne>40, 4-10 times that of traditional materials, Figure 1C), and the size of Me is positively correlated with side chain length, indicating precise control of Me through side chain modification, while the high Ne value directly explains the material’s excellent high-temperature stability.
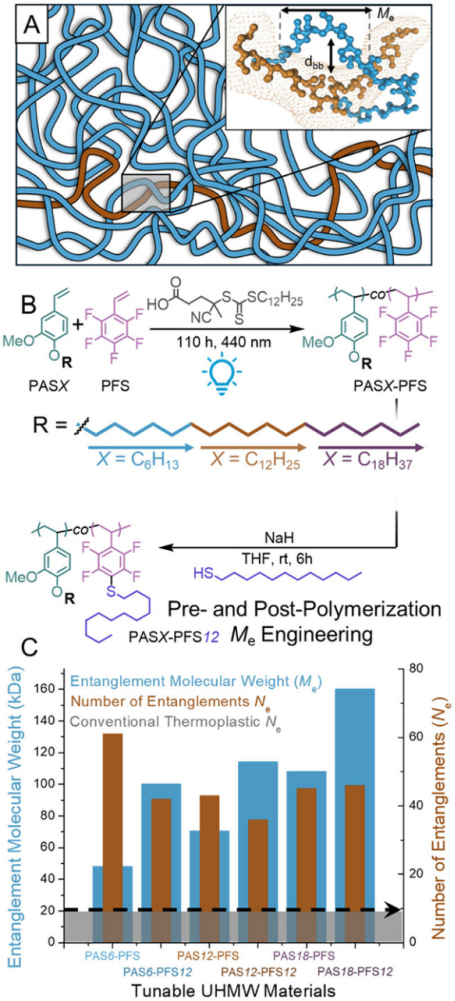
Figure 1. (A) Schematic diagram of the entangled structure in the polymer melt.(B) Synthesis steps for polymers with tunable entanglement molecular weight (Me) through photoinitiated polymerization and post-polymerization modification.(C) The Me and Ne of the high-density copolymer in this study, with its Ne exceeding that of traditional thermoplastics (gray area) by 3-10 times.
Through dynamic mechanical analysis (DMA), the effect of temperature changes on modulus was characterized. The results showed that the UHMW PASX-PFS samples maintained a rubber-like modulus at 200°C (far above Tg,DSC) while the low molecular weight (LMW) samples (Ne<10, Mn<500 kDa) flowed at a temperature 30°C higher than Tg,DMA (Figure 2A). Both had similar Tg,DMA and Me values (Figure 2A, D, E), indicating no significant differences in chemical composition, suggesting that the high-temperature thermomechanical properties of the UHMW samples stem from their high Ne value characteristics.
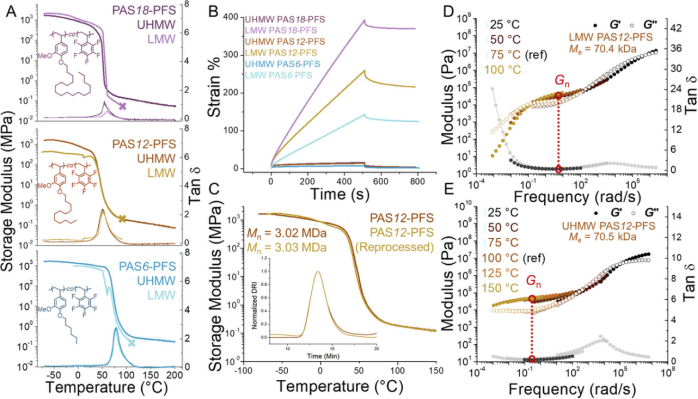
Figure 2. (A) Dynamic mechanical analysis (DMA) of ultrahigh molecular weight polystyrene polymers compared to low molecular weight (LMW, <500 kg mol−1) samples.(B) Strain-recovery experiments show that ultrahigh molecular weight polymers can resist permanent deformation, while low molecular weight samples undergo permanent deformation.(C) Comparison of DMA before and after reprocessing of ultrahigh molecular weight PAS12-PFS materials. Insets: Size exclusion chromatography (SEC) refractive index distribution curves of samples before and after processing.(D) Low molecular weight PAS12-PFS small amplitude oscillatory shear rheology master curve at 75°C.(E) Ultrahigh molecular weight PAS12-PFS 100°C small amplitude oscillatory shear rheology master curve.
The thermal stability of the UHMW samples, which resembles a chemically crosslinked network at high temperatures, does not arise from covalent bonds. The study found that these polymers can be repeatedly melted or solution processed, and their thermomechanical properties (Figure 2C) and chemical structure (Figure 2C inset) remain unchanged.This is something traditional covalent crosslinked networks cannot achieve, highlighting the importance of achieving UHMW in thermochemically stable polystyrene-like polymers.
UHMW PASX-PFS exhibits excellent creep resistance due to its ultralong relaxation time. The authors employed an improved creep testing method (2500 Pa constant stress, 500 seconds loading +300 seconds recovery, Figure 2B) and found that the UHMW samples’ resistance to deformation and recovery ability significantly outperformed that of the LMW samples and other high-performance adaptive networks, highlighting the unique advantage of UHMW materials in maintaining dimensional stability. These findings indicate that UHMW polymers achieve lasting thermomechanical stability through tunable Me values, combining excellent properties that traditional thermoplastics and covalent network materials cannot simultaneously obtain.
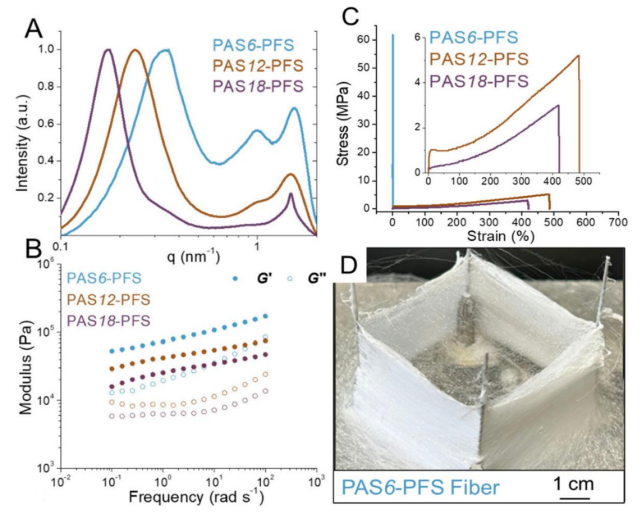
Figure 3. (A) X-ray scattering measurements of ultrahigh molecular weight PASX-PFS copolymers.(B) Small amplitude oscillatory shear rheology measurements of ultrahigh molecular weight PASX-PFS at 125°C.(C) Stress-strain curves show that ultrahigh molecular weight PAS12-PFS and PAS18-PFS exhibit strain hardening above the glass transition temperature (Tg) (inset shows high strain region).(D) Photos of PAS6-PFS fibers prepared by spinning.
By regulating the entanglement molecular weight (Me), the modulus and snake-like dynamics of UHMW polymers can be effectively altered. The X-ray scattering data (Figure 3A) shows that the main chain spacing significantly increases with the growth of the para-alkoxy side chains (0.17–0.34 Å), while the side chain spacing remains constant (4-4.2 Å), but the PAS18 units pack more orderly. These X-ray scattering features correlate with small amplitude oscillatory shear (SAOS) rheology test results (Me=ρRT/G0N∝dbb2), which shows that as the snake-like diameter of the UHMW chains increases, the storage modulus G’ and loss modulus G” both decrease (Figure 3B).
Due to the differences in relaxation dynamics between the main chain and side chains, UHMW polymers exhibit unique multi-stage response characteristics (Figure 3C): PAS6-PFS (Tg=55°C) exhibits high-strength glassy state (65.7 MPa, 1.02% strain), while PAS12/18-PFS (Tg/Tm<35°C) demonstrates elastomeric behavior, with high ductility (361-449% strain) and strain hardening characteristics (4.57-6.67 MPa). This structural advantage can be directly translated into processing performance, successfully producing self-supporting fiber mats through spinning processes (3000 rpm), providing a new paradigm for developing high-performance fibers with tunable modulus.
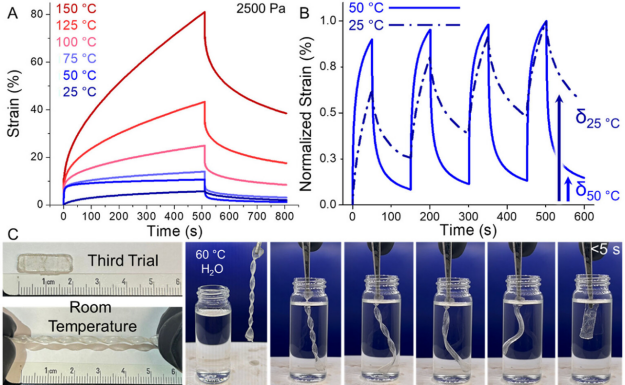
Figure 4. (A) PAS12-PFS material’s temperature rise creep recovery experiment.(B) Creep cycle tests indicate that the material’s δ value is higher after four cycles at 25°C compared to 50°C.(C) PAS12-PFS rods formed a helical structure after two room temperature deformations, fully recovering within 5 seconds when heated above Tg .
Differences in relaxation dynamics between the ultralong polymer main chains and their oligomeric side chains endow these materials with excellent shape memory properties. When the deformation temperature is slightly below or equal to its Tg , the amount of permanent deformation produced by the material is significantly higher than that at slightly above Tg temperature (Figure 4A ). For example, with PAS12-PFS (Tg=22°C), the accumulated permanent deformation reached 2.2% after four creep cycles at 25°C, while at 50°C it was only 1.3% (Figure 4B ). The authors believe that the differences in relaxation temperatures between the main and side chains lead to optimal strain recovery in intermediate temperature ranges, indicating that this material possesses unique low-temperature programmable shape memory characteristics.
To verify this hypothesis, the authors stretched the compressed molded PAS12-PFS strips at room temperature, twisting them into a composite deformation and maintaining the temporary shape. When immersed in a water bath above 60°C (above Tg), the samples rapidly recovered their original shape within 5 seconds. The authors believe that the kinetic stability of this temporary helical shape arises from a triple mechanism:(1) low-temperature interlocking between side chains, (2) physical constraints of the main chain entangled network, (3) restricted segment motion below Tg . These results indicate that by regulating the relaxation dynamics of UHMW polymers, room temperature shape memory functionality can be achieved, a characteristic that is difficult to design in traditional thermoplastics.
Furthermore, the authors introduced dodecyl side chains through post-polymerization modification (PPM) (Figure 1B), significantly enhancing the Me value and chain rigidity of PASX-PFS, resulting in significant softening of the material. X-ray scattering showed that the main chain spacing of PAS12-PFS remained basically unchanged, while the spacing of PAS6/18-PFS changed significantly (Figure 5A ). Although the modulus of the modified material decreased, it still maintained a high entanglement characteristic, yielding only above Tg 100℃ (Figure 5B ), and the strain increased in the creep test at 125℃, but the recovery rate remained high (Figure 5C ).
In terms of mechanical properties, the modified materials exhibited significantly enhanced ductility: PAS6-PFS transitioned from brittle fracture (65 MPa,1.2% ) to PAS6-PFS12 ultra-soft elastomer (0.04 MPa,>650% elongation); PAS18-PFS12 exhibited even greater performance with >1400% elongation and >90% recovery rate (Figure 5D ).
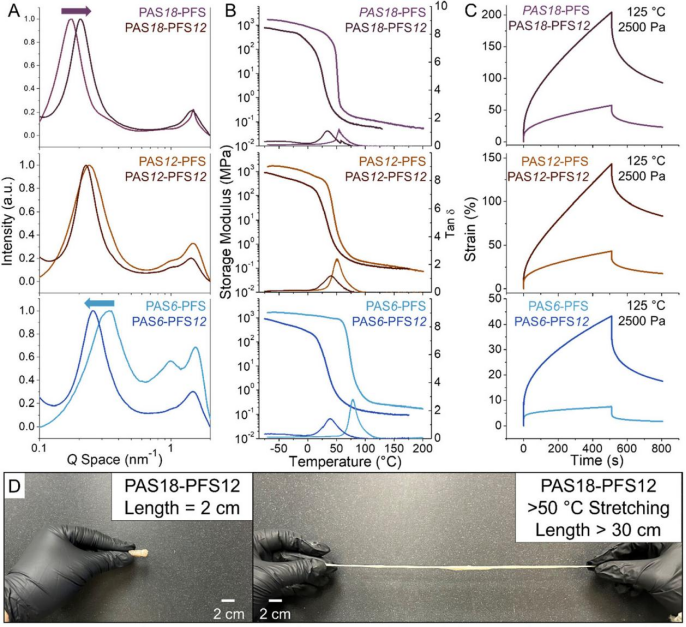
Figure 5. (A) X-ray scattering tests of PASX-PFS before and after modification with dodecanethiol.(B) Dynamic mechanical analysis comparison of PASX-PFS and PASX-PFS12.(C) Creep experiments conducted at 125°C and 2500 Pa pressure.(D) PAS18-PFS12 can undergo elastic deformation when heated above the glass transition temperature.
This study developed a controllable ultrahigh molecular weight polystyrene polymer system with high entanglement density through photoinitiated transfer termination polymerization techniques. This material combines excellent processing performance with high-temperature dimensional stability. By employing molecular-level side chain modification strategies, precise control of Me values and broad adjustment of mechanical properties were achieved. The study confirms that its tunability aligns with snake-like model theory and successfully developed ultra-soft materials with room temperature shape memory effects, breaking the performance limits of traditional amorphous thermoplastics. This work provides a new paradigm for precisely regulating polymer entanglement networks through chemical design.
Authors:XJQ
DOI: 10.1021/jacs.5c08357
Link: https://doi.org/10.1021/jacs.5c08357
Previous article