
 Follow us to receive such great articles every day!
Follow us to receive such great articles every day!
WeChat ID:kqjy360

Authors: Tang Xiaoyu, Gu Dongkun, Li Dong, Niu Qianyun, Nanyang Dental Hospital; Wu Dandan, Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Dental School
Oral-Facial-Digital Syndrome (OFDS) is a unique developmental genetic disorder characterized by malformations of the mouth, face, and fingers, with multiple subtypes. Among them, Oral-Facial-Digital Syndrome Type I (OFD1) is the most common type, also known as Papillon-Leage-Psaum syndrome, first reported by Papillon-Leage and Psaum in 1954. It is an X-linked dominant inheritance disorder, often accompanied by brain structural abnormalities and cystic diseases of other organs in addition to oral, facial, and finger malformations.
The incidence of OFD1 is approximately 1 in 250,000 to 1 in 50,000, with 75% being sporadic cases. Currently, only 16 cases have been reported in China. From November 2015 to September 2019, the Department of Oral and Maxillofacial Surgery at Nanyang Dental Hospital treated 5 patients with OFD1, and the findings are summarized as follows.
1. Cases and Methods
1.1 Clinical Data Review
From November 2015 to September 2019, the Department of Oral and Maxillofacial Surgery at Nanyang Dental Hospital collected case data from 5 patients with OFD1, all of whom were female, including 2 mother-daughter pairs.
1.1.1 Group 1 Patients
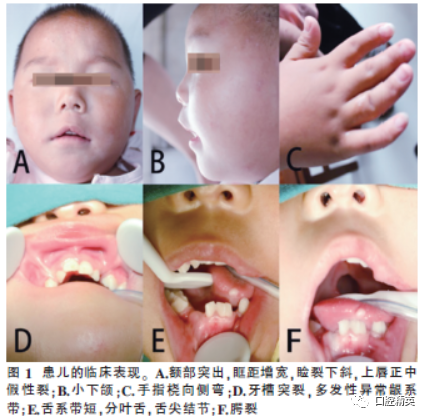
Patient, 3 years old, full-term birth. Clinical manifestations (Figure 1): ① Sparse hair, symmetrical facial features, prominent forehead, widened interorbital distance, downward slant of palpebral fissures, pseudo-cleft in the midline of the upper lip, and downward arched oral cleft; ② Normal mouth opening, short lingual frenulum, lobulated tongue, nodules on the tongue tip, multiple abnormal gingival frenula, incomplete cleft palate, bilateral maxillary and mandibular alveolar ridge clefts, primary dentition, missing bilateral mandibular lateral incisors and maxillary primary cuspids; ③ Ulnar deviation of the index finger, radial deviation of the 4th and 5th fingers; ④ Doppler ultrasound showed no polycystic kidney.
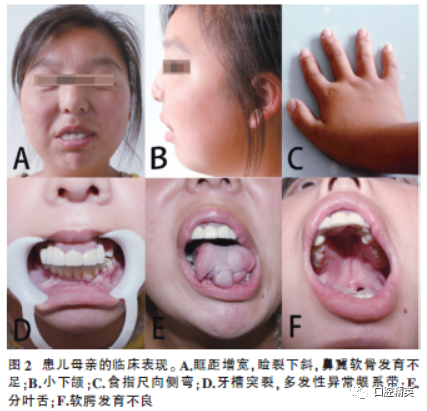
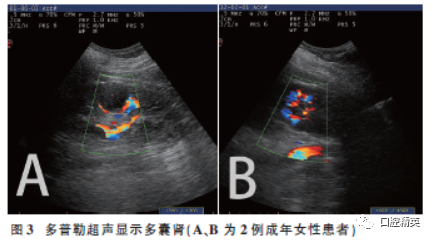
The mother of the patient, 29 years old, clinical manifestations (Figure 2): ① Sparse hair, asymmetrical facial features, prominent forehead, widened interorbital distance, downward slant of palpebral fissures, underdeveloped nasal cartilage, downward arched oral cleft, small mandible; ② Lobulated tongue, multiple abnormal gingival frenula, underdeveloped soft palate, bilateral maxillary and mandibular alveolar ridge clefts, dental arch defects, denture restoration; ③ Short fingers, radial deviation of the index finger; ④ Doppler ultrasound: showed polycystic kidney (Figure 3A). ⑤ Non-consanguineous marriage, no similar cases in the family, and a history of one induced abortion.
1.1.2 Group 2 Patients
Patient, 2 years old, full-term birth. Clinical manifestations: ① Basically symmetrical face, prominent forehead, widened interorbital distance, downward arched oral cleft. ② Short lingual frenulum, lobulated tongue, nodules on the tongue tip, multiple abnormal gingival frenula, attached to the alveolar ridge apex, bony depression between the left maxillary lateral incisor and second premolar, right mandibular lateral incisor not erupted, left mandibular primary lateral incisor erupted, abnormal frenulum attached to the mesial of the first mandibular primary molar, bilateral maxillary and mandibular alveolar ridge clefts, incomplete cleft palate. ③ No abnormalities of fingers or toes. ④ Doppler ultrasound showed no polycystic kidney.
The mother of the patient, 31 years old, main manifestations: ① Asymmetrical facial features, prominent forehead, widened interorbital distance, underdeveloped nasal cartilage, downward corners of the mouth, downward arched oral cleft, miliaria; ② Short lingual frenulum, lobulated tongue, nodules on the tongue tip, multiple abnormal gingival frenula in the upper and lower jaws; ③ Doppler ultrasound showed polycystic kidney (Figure 3B); ④ No abnormalities of fingers or toes; ⑤ Non-consanguineous marriage, no similar cases in the family, and a history of three induced abortions.
1.1.3 Group 3 Patients
Patient, 2 years old, clinical manifestations: ① Basically symmetrical face, prominent forehead, widened interorbital distance, flat nasal tip; ② Lobulated tongue, nodules on the tongue tip, short lingual frenulum, multiple abnormal gingival frenula, missing bilateral mandibular primary lateral incisors, alveolar ridge clefts, incomplete cleft palate; ③ Ulnar deviation of the index finger; ④ Doppler ultrasound showed no polycystic kidney; ⑤ The patient was adopted, and family history is unclear. None of the 5 patients exhibited intellectual or hearing impairments.
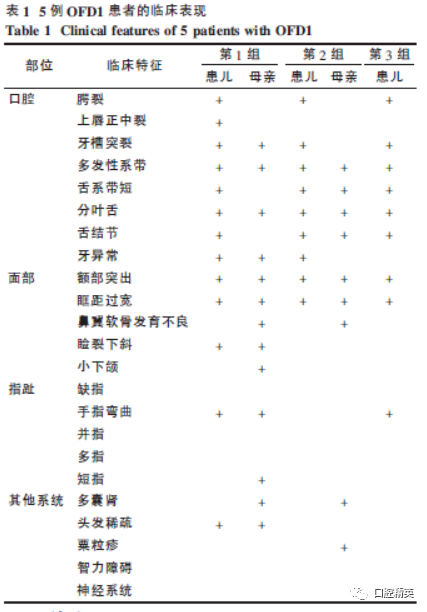
All clinical features are summarized in Table 1.
1.2 Treatment
After admission, patients underwent physical examination, routine laboratory tests, chest X-ray, electrocardiogram, and abdominal ultrasound to exclude absolute contraindications for surgery. Under general anesthesia, palatoplasty (3 cases), excision of tongue nodules (4 cases), correction of lobulated tongue deformity (5 cases), correction of lingual frenulum (4 cases), correction of gingival frenulum (5 cases), and vestibular groove deepening (5 cases) were performed.
2. Results
Based on the patients’ medical history and clinical features, all 5 patients were diagnosed with OFD1. Two of the mothers had polycystic kidney disease, while three of the pediatric patients showed no signs of polycystic kidney. There were no unexpected events or complications during the perioperative period. Postoperatively, the tongue morphology was good, with normal protrusion, good morphology of the gingival frenulum and vestibular groove, good healing of the cleft palate incision, and normal soft palate mobility and velopharyngeal closure function. The pathological report of the tongue nodules indicated a lingual hamartoma (Figure 4).
3. Discussion
3.1 Classification and Etiology
Oral-Facial-Digital Syndrome was first reported by Mohr in 1941. Due to its complex clinical manifestations, there are 14 classifications, with 5 types still not clearly defined. However, Bruel et al. suggested that OFDS can be classified into three main subtypes: OFD1, OFD4, and OFD5 based on clinical and mutation correlations. Current genetic studies have confirmed that OFD1 is an X-linked dominant inheritance disorder, with the pathogenic gene Cxorf5 located on Xp22. To date, 128 gene mutations have been identified in OFD1, with 50% being frameshift mutations. This random inactivation and individual variation can explain the diversity of clinical manifestations in OFD1. The pathogenesis of the clinical features of OFD1 is related to primary ciliary dysfunction, and abnormal migration and differentiation of cranial neural crest cells may be the pathological cause of the oral and maxillofacial defects in OFDS.
3.2 Clinical Features of OFD1
The clinical features of OFD1 include: ① Oral deformities: common multiple abnormal gingival frenula and alveolar ridge clefts, abnormal frenula extending from the buccal mucosa to the alveolar ridge, causing alveolar ridge depression; over 50% are accompanied by varying degrees of cleft palate; lobulated tongue, short lingual frenulum, approximately 30% present with lingual hamartomas; dental deformities include congenital missing teeth (most common), supernumerary teeth, enamel hypoplasia, dental arch deformities, and occlusal disorders. ② Facial deformities: pseudo-cleft in the midline of the upper lip, small mandible, prominent forehead, widened interorbital distance (approximately 33%), downward slant of palpebral fissures, underdeveloped nasal cartilage (approximately 65%). ③ Digit abnormalities (12%): common short fingers, syndactyly, and ulnar deviation of other fingers, polydactyly (50%), and supernumerary digits (1%-2%). ④ Structural brain abnormalities (up to 65%): including intracranial cysts, agenesis of the corpus callosum, and cerebellar hypoplasia, especially those with cerebellar atrophy may have seizures and ataxia, approximately 50% may have mild intellectual disability or learning difficulties. ⑤ Polycystic kidney disease (PKD), patients may also have cysts in the liver, pancreas, and ovaries. ⑥ Others: otitis media, hair abnormalities (approximately 20%), miliaria (10%), congenital heart disease.
This group of cases confirms the complexity and variability of the OFD1 phenotype. Affected individuals do not exhibit all clinical features of OFD1, but oral manifestations are relatively consistent. The two adult females both presented with PKD, consistent with previously reported incidence rates.
3.3 Diagnosis and Differential Diagnosis
The main diagnostic criteria for OFD1 include: ① unique oral, facial, and digital abnormal phenotypes; ② characteristic X-linked dominant inheritance pattern with male lethality; ③ polycystic kidney; ④ molecular genetic testing. Although there have been reports of male patients, these males are considered malformed fetuses of affected females. Additionally, PKD has been confirmed as a characteristic disease distinguishing OFD1 from other subtypes, being the only significant manifestation in affected females, but often appears in late childhood or adulthood. Most patients can be identified through sequencing for Cxorf5 mutations, but 20% of patients still cannot detect genetic variations.
Our group of patients did not undergo relevant genetic testing due to family refusal. OFD1 needs to be differentiated from other subtypes, mainly OFD2, which is autosomal recessive and shares many common features with OFD1, such as cleft tongue, lingual hamartomas, midline cleft of the upper lip, multiple frenula, curved fingers, syndactyly, and polydactyly, but commonly presents with bilateral thumb polydactyly, hearing impairment, and can have brain malformations and hydrocephalus, atrioventricular canal defects, and endocardial cushion defects, without polycystic kidney, miliaria, alveolar clefts, and hair abnormalities. OFD8 and OFD9, although X-linked dominant, are characterized by polydactyly, tibial and radial defects, epiglottis abnormalities, and retinal anomalies, and midline cleft lip.
3.4 Treatment
The treatment of OFD1 is recommended to be a multidisciplinary comprehensive sequential treatment primarily based on surgery. In addition to routine examinations, attention should also be paid to brain, abdominal organs, heart, and other system examinations and functional assessments. Close monitoring is required during and after general anesthesia to prevent serious complications such as aspiration, upper airway obstruction, and organ dysfunction. Surgical treatments include repair of cleft lip and palate, correction of abnormal frenula, excision of lingual hamartomas, and reconstruction of lobulated tongue. Severe alveolar ridge clefts may require bone grafting surgery at ages 9-11.
Due to the common absence of lateral incisors in children, temporary space maintainers may be used, and orthodontic treatment can be performed after the replacement period. For adult patients with dental arch defects and occlusal disorders, denture restoration and orthodontic treatment may be performed. For patients with cleft palate, speech therapy may be necessary, and hearing assessments should be conducted; those with otitis media should receive active and appropriate treatment to avoid serious complications such as tympanic membrane perforation. Most PKD patients have severe renal insufficiency, leading to end-stage renal disease (71%), with ages ranging from 11 to 70 years, requiring hemodialysis or peritoneal dialysis, or even kidney transplantation. Although not all patients in this group exhibited PKD, the high incidence of PKD warrants regular monitoring of renal function.
Patients with central nervous defects and seizures should be actively controlled to reduce seizures and avoid further brain damage from prolonged recurrent seizures. Syndactyly, polydactyly, and finger curvature should be surgically corrected to restore hand function and aesthetics. Those with learning and cognitive disabilities require special education. 3.5 Genetic Risk Based on its inheritance pattern and etiology, the risk of healthy mothers of OFD1 patients giving birth to another OFD1 daughter is less than 1%, while the risk of OFD1 female offspring is 50%. Prenatal testing and genetic diagnosis should be provided for high-risk pregnancy populations, along with genetic counseling.
In conclusion, OFD1 is clinically rare, and it is crucial to distinguish it from other subtypes of OFDS. While treating oral and maxillofacial deformities, attention should also be paid to the examination of other systemic defects to guide the medical management and health monitoring of these patients, and to provide accurate genetic counseling and advice.
