In the 2008 (4th edition) World Health Organization (WHO) classification, this disease category was first established, originally named myeloid/lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1 genes. In the revised 4th edition WHO classification, PCM1::JAK2 and its genetic variants were introduced as provisional entity subtypes within this category. In the current classification, this category has been further expanded to include FLT3 rearrangements and t(9;12)(q34;p13)/ETV6::ABL1 as new members, and PCM1::JAK2 and its genetic variants (ETV6::JAK2 and BCR::JAK2) have been elevated to formal entity subtypes. Additionally, the category name has been changed to myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions (M/LN-eo-TK) to emphasize that the molecular genetic alterations underlying these hematologic tumors lead to constitutive tyrosine kinase (TK) signaling, making targeted therapy acceptable. These tumors originate from mutated multipotent stem cells in the bone marrow and may differentiate along myeloid and/or lymphoid lineages, often resulting in complex and diverse clinical manifestations. Although eosinophilia is frequently observed, it is not always present; retaining eosinophilia in the category name helps raise suspicion for these gene-defined lesions, especially in cases with cryptic rearrangements. Other potential candidate genes for M/LN-eo-TK include but are not limited to rearrangements involving LYN, FGFR2, NTRK3, RET, SKY, ALK, and KIT. So far, such cases have been limited to individual case studies, and more data are needed to understand their clinicopathological features, overlap with other gene-defined entities, and responses to tyrosine kinase inhibitor (TKI) therapy. However, for cases presenting as chronic phase or blast phase myeloid neoplasms, or hematologic tumors involving both myeloid and lymphoid lineages, often accompanied by eosinophilia and responsive to TKI therapy, a diagnosis of M/LN-eo-TK is appropriate. With the widespread application of RNA sequencing in clinical practice, more tyrosine kinase gene fusions may be discovered and added to this evolving disease category. An important note is that the diagnosis of M/LN-eo-TK applies to hematologic tumors that carry specific genetic abnormalities at the time of initial presentation, not to cases that acquire such abnormalities (rarely) during disease progression/evolution.
M/LN-eo-TK (possibly referring to myeloid/lymphoid neoplasms involving tyrosine kinase genes, specifically named here) can present as B-cell or T-cell acute lymphoblastic leukemia/lymphoma (ALL). BCR::ABL1-like B-cell acute lymphoblastic leukemia (B-ALL), de novo T-cell acute lymphoblastic leukemia (T-ALL), or even mature T-cell lymphoma may involve one of the tyrosine kinase (TK) genes. These differences can be attributed at least in part to the different partner genes involved in the tyrosine kinase gene fusions. For example, t(5;12)(q32;p13.2)/ETV6::PDGFRB fusion genes typically present with multilineage involvement, which is a characteristic feature of M/LN-eo-TK; whereas when PDGFRB fuses with partner genes such as EBF1, SSBP2, TNIP1, ZEB2, and ATF7IP, it usually presents as de novo B-cell acute lymphoblastic leukemia without myeloid involvement, and should be diagnosed as BCR::ABL1-like B-ALL. Similarly, fusions of the JAK2 gene with certain partner genes (such as t(5;9)(q14.1;p24.1)/STRN3::JAK2 fusion gene and PAX5::JAK2 fusion gene) are typically seen in BCR::ABL1-like B-ALL, which by definition is not M/LN-eo-TK. Complicating matters, certain fusion partner genes (such as ETV6::JAK2 and ETV6::ABL1) can be observed in both disease entities. This overlap phenomenon exists across all tyrosine kinase genes, and diagnosis should follow general principles: for M/LN-eo-TK cases that initially present as B-cell or T-cell acute lymphoblastic leukemia, the tyrosine kinase gene fusion should involve both myeloid and lymphoblasts. In such cases, the chronic myeloid neoplasm (CMN) component of M/LN-eo-TK may appear before, during, or after treatment for acute lymphoblastic leukemia (Figure 6.1). This concept is similar to the distinction between chronic myeloid leukemia (CML) presenting as acute transformation and de novo adult BCR::ABL1-positive acute lymphoblastic leukemia. For practical purposes, fluorescence in situ hybridization (FISH) technology to detect the corresponding gene fusions in a defined myeloid cell population helps distinguish M/LN-eo-TK from acute lymphoblastic leukemia.
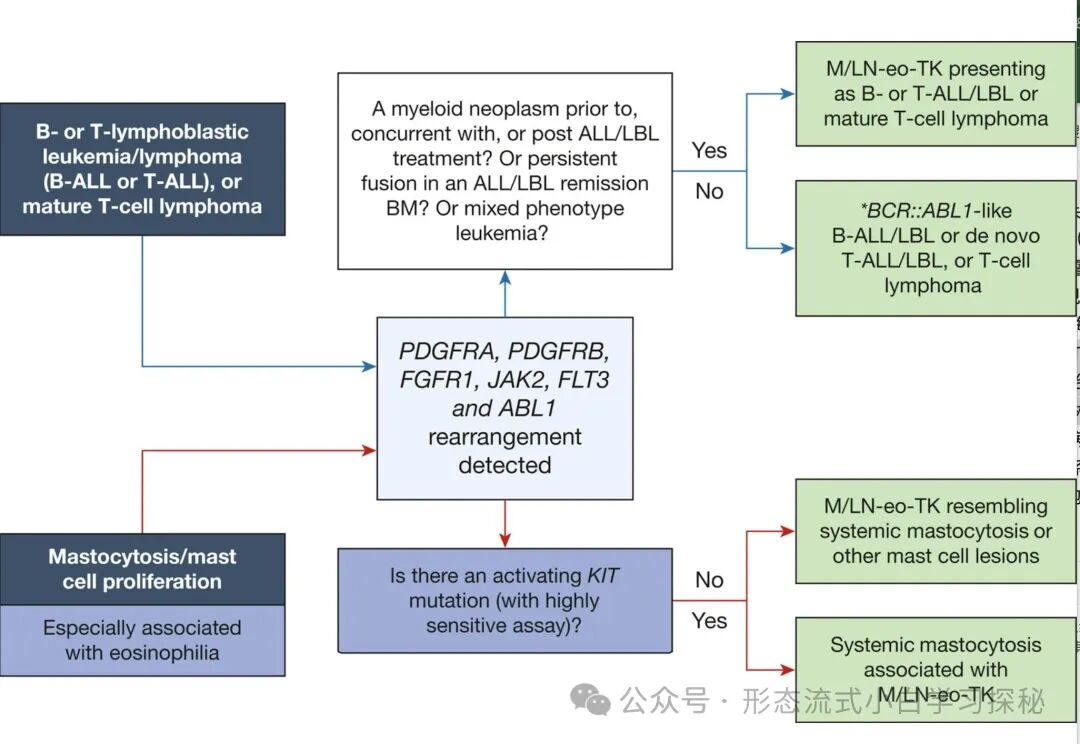
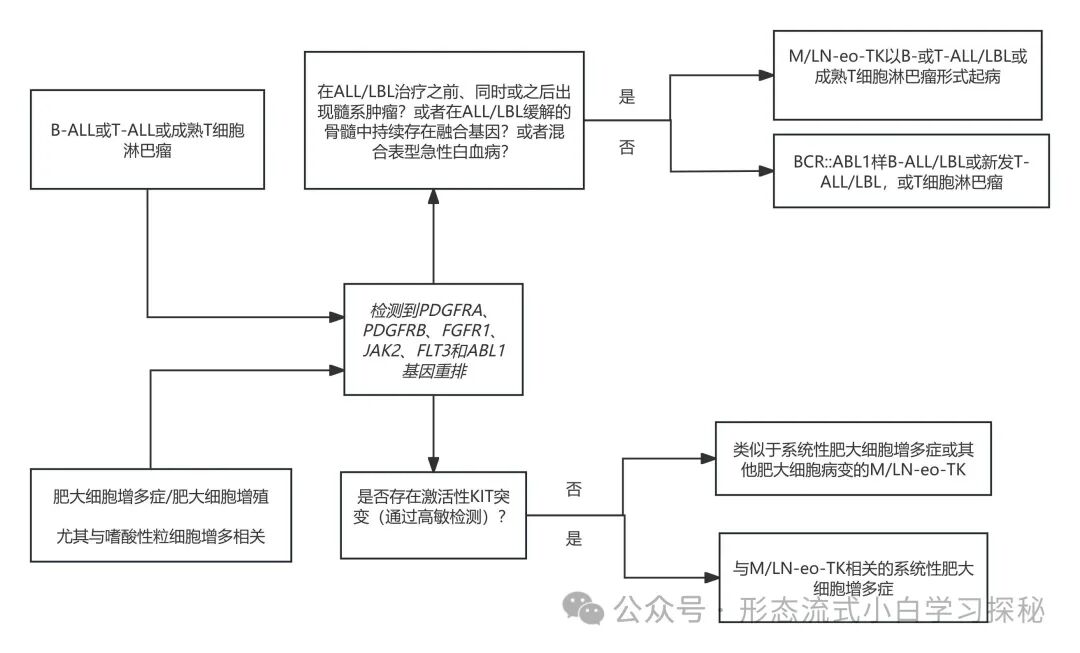
Figure 6.1 illustrates overlapping disease entities. (1) Differentiation between myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase fusions (M/LN-eo-TK) and BCR::ABL1-like B-cell acute lymphoblastic leukemia/lymphoma (B-ALL/LBL), de novo T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/LBL), or mature T-cell lymphoma; (2) Differentiation between myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase fusions (M/LN-eo-TK) and systemic mastocytosis associated with M/LN-eo-TK. Certain partner genes, such as those fused with PDGFRB like EBF1, SSBP2, TNIP1, ZEB2, and ATF7IP; and those fused with JAK2 like ATF7IP, EBF1, GOLGA5, HMBOX1, OFD1, PAX5, PPFIBP1, RFX3, SMU1, SNX29, SSBP2, STRN3, TERF2, TPR, USP25, ZBTB46, ZNF274, and ZNF340, are almost exclusively reported in BCR::ABL1-like B-cell acute lymphoblastic leukemia, and not seen in M/LN-eo-TK.
As previously mentioned, abnormal mast cells are often observed in myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo-TK). In rare cases, especially in those with elevated serum tryptase levels, mast cells may proliferate extensively, resembling systemic mastocytosis (SM). However, these cases never exhibit the KIT D816V mutation, and the response to tyrosine kinase inhibitor (TKI) therapy is similar to that of other myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo-TK). In the current classification, if one of the tyrosine kinase (TK) gene fusions is detected, such cases are classified as myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo-TK). For suspected cases of systemic mastocytosis (SM) with eosinophilia but without KIT mutations, it is strongly recommended to use high-sensitivity molecular detection methods to check for TK gene fusions. In rare cases, both TK gene fusions and KIT D816V mutations may coexist, which may represent two diseases with independent clones occurring simultaneously, or a subclone with acquired activating KIT mutations, both of which fall under the category of systemic mastocytosis-associated myeloid neoplasms (SM-AMN), where myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo-TK) are the “associated myeloid neoplasms (AMN).”
The key differential points for these overlapping disease entities are illustrated in Figure 6.1.
1. Myeloid/lymphoid neoplasms with eosinophilia and PDGFRA rearrangements
Definition
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) stem cells with PDGFRA gene rearrangements or, in rare cases, due to activating mutations leading to constitutive activation of tyrosine kinase (TK). Most cases carrying the FIP1L1::PDGFRA fusion gene (approximately 90%) present as chronic myeloid neoplasms (CMN) with eosinophilia, while a minority of cases exhibit other different fusion gene partners.
Synonyms: None
Epidemiology
This disease shows a significant male predominance, with a male-to-female ratio of approximately 17:1. The median age of onset is in the late 40s, with a peak age range of 25 to 55 years. This is the most common type of gene rearrangement in myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene rearrangements (M/LN-eo-TK).General Clinical Features
Patients may present with fatigue, shortness of breath, and systemic symptoms, with some experiencing pruritus, respiratory symptoms, and rare cardiac or gastrointestinal symptoms. The latter symptoms may be related to end-organ damage caused by eosinophilic infiltration or enzymes and other proteins released by eosinophils. Splenomegaly is common, seen in 40% to 50% of patients, with some also exhibiting hepatomegaly. Some patients may present with extramedullary disease (EMDs). 90% of patients have eosinophilia, and almost all patients carrying the FIP1L1::PDGFRA fusion gene will exhibit eosinophilia, although some patients may not meet the criteria for eosinophilia (1.5 × 10⁹/L). Some patients may present with anemia, thrombocytopenia, and monocytosis. Other laboratory features include significantly elevated serum vitamin B12 and increased serum tryptase levels.
Morphology and Immunophenotype
Eosinophilia in peripheral blood is common, with most showing mature forms. In over 50% of cases, significant eosinophilic abnormalities can be observed, including reduced granules and/or uneven granule size, abnormally large or small granules, cytoplasmic vacuolation, and excessive or insufficient nuclear lobulation, as well as increased eosinophil volume (Figure 6.2). The counts of blast cells, basophils, and monocytes typically do not increase, but may be seen in some cases with partners other than FIP1L1.

Figure 6.2 shows common manifestations of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene rearrangements (M/LN-eo-TK) in peripheral blood and bone marrow, illustrated using cases of PDGFRA (A, C, E, F) and PDGFRB (B, D, F, H). Peripheral blood often shows eosinophilia (A and B); eosinophilic abnormalities are common, such as insufficient (A) or excessive (B) nuclear lobulation, abnormal granules (A and B), and larger morphology (A). Some cases may also show granulocyte abnormalities, such as abnormal granulocyte development and the appearance of granulocyte precursors (A). Eosinophilic abnormalities in the bone marrow are usually less pronounced than in peripheral blood but often show increased eosinophilic precursors. Monocytosis may be seen in 15% to 20% of PDGFRB rearrangement cases; in this case, it can also be observed in the bone marrow (D). Bone marrow biopsy typically shows significantly active cellular proliferation (E-G); megakaryocytes may be significantly reduced (E), or appear large with distant nuclear spacing (F), or as small, abnormally developed forms (G). Scattered spindle-shaped mast cells are commonly observed and can be seen in any M/LN-eo-TK (H). (A-D: Wright-Giemsa stain; E-G: Hematoxylin-Eosin stain; H: tryptase stain; A-D: 100×, E: 20×, F-H: 40×).
Bone marrow cellular proliferation is significantly active, with a marked increase in eosinophils. Eosinophils are primarily mature forms, but in some cases, immature eosinophilic precursors may show a left shift. Eosinophilic abnormalities (if present) are usually not as pronounced as in peripheral blood. Megakaryocytes are often reduced, but may also be normal or increased, with morphology showing no abnormalities, or presenting as large, multilobulated megakaryocytes, small abnormal forms, or various morphologies (Figure 6.2). Focal necrosis and Charcot-Leyden crystals may be observed. Mast cells are often increased and spindle-shaped, mostly distributed sparsely or forming loose, non-cohesive clusters, but in some cases, mast cell proliferation may form aggregates resembling systemic mastocytosis (SM). Mild bone marrow fibrosis is common, but MF-2 or MF-3 grade fibrosis is only seen in 20% of cases.
A minority of patients present with acute leukemia, such as acute myeloid leukemia (AML) or T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/LBL), which may have previously been associated with eosinophilia. Extramedullary infiltration or tumor lesions are not uncommon, often involving lymph nodes, epidural and/or paravertebral spaces, bones, and skin tissues. These lesions are usually of myeloid origin, with cell morphology ranging from immature to mature; in some cases, they may be T-ALL/LBL, rarely B-ALL/LBL. The presence of eosinophils varies. These extramedullary disease (EMD) lesions may appear at initial diagnosis or during disease progression. If there is an increase in blast cells or a biopsy of EMD lesions, flow cytometric immunophenotyping and/or immunohistochemical methods should be used to determine their phenotypic characteristics, with the presence of abnormal mast cells often aiding in confirming clonal eosinophilia.
Genetics
The PDGFRA gene rearrangement is usually due to an approximately 800 kb interstitial deletion in the q12 region of chromosome 4 (including the CHIC2 gene), leading to the fusion of FIP1L1 with the PDGFRA gene. This rearrangement is difficult to detect in routine chromosomal karyotype analysis and requires identification through fluorescence in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT-PCR), or RNA sequencing (RNAseq) techniques. To date, seven other partner genes have been reported to rearrange with PDGFRA, including CDK5RAP2/ins(9;4)(q33;q12q25), ETV6/t(4;12)(q12;p13), FOXP1/t(3;4)(p13;q12), KIF5B/t(4;10)(q12;p11), STRN/t(2;4)(p22;q12), TNKS2/t(4;10)(q12;q23), and BCR/t(4;22)(q12;q11) (see Table 6.1). These gene variants are usually not difficult to detect, but confirming the involvement of PDGFRA in the fusion is crucial. For example, it has been reported that some acute myeloid leukemia (AML) cases with t(4;12)(q12;p13) translocation detected PDGFRA gene rearrangement by FISH, but were insensitive to imatinib treatment. An RNA sequencing study showed that in the 4q12 region, the ETV6 gene fused with SCFD2, CHIC2, and GSX2, and these fusion genes are too close to the PDGFRA gene location to be distinguished by chromosomal analysis or FISH analysis (leading to false-positive FISH results). Notably, t(4;12)(q12;p13) translocations carrying true ETV6::PDGFRA fusion genes typically present as chronic myeloid neoplasms (CMN) with eosinophilia, while other partner genes in the 4q12 region (non-PDGFRA) involved in t(4;12)(q12;p13) translocations usually do not present with eosinophilia. Additionally, false-negative FISH results may occur, indicating that in highly suspected cases, further genetic testing (such as RNA sequencing or optical genome mapping) is needed. It has been reported that mutations exist in 20% to 50% of myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo) cases with PDGFRA gene rearrangements, including ASXL1, BCOR, DNMT3A, ETV6, SRSF2, PTPN11, and RUNX1. Currently, the significance of these additional mutations remains largely unclear.
Table 6.1 Genetic abnormalities, clinical presentations, and targeted therapy of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions (M/LN-eo-TK)
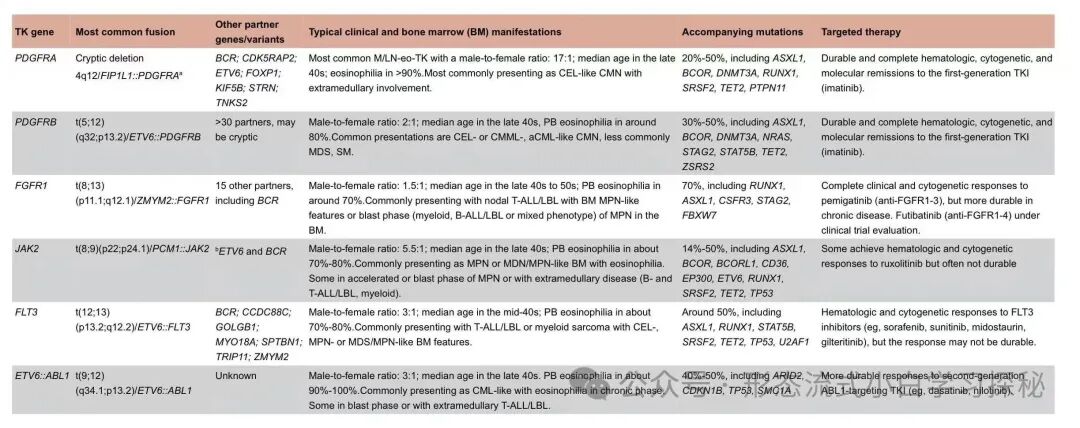
Table 6.1 Genetic abnormalities, clinical presentations, and targeted therapy of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions (M/LN-eo-TK)
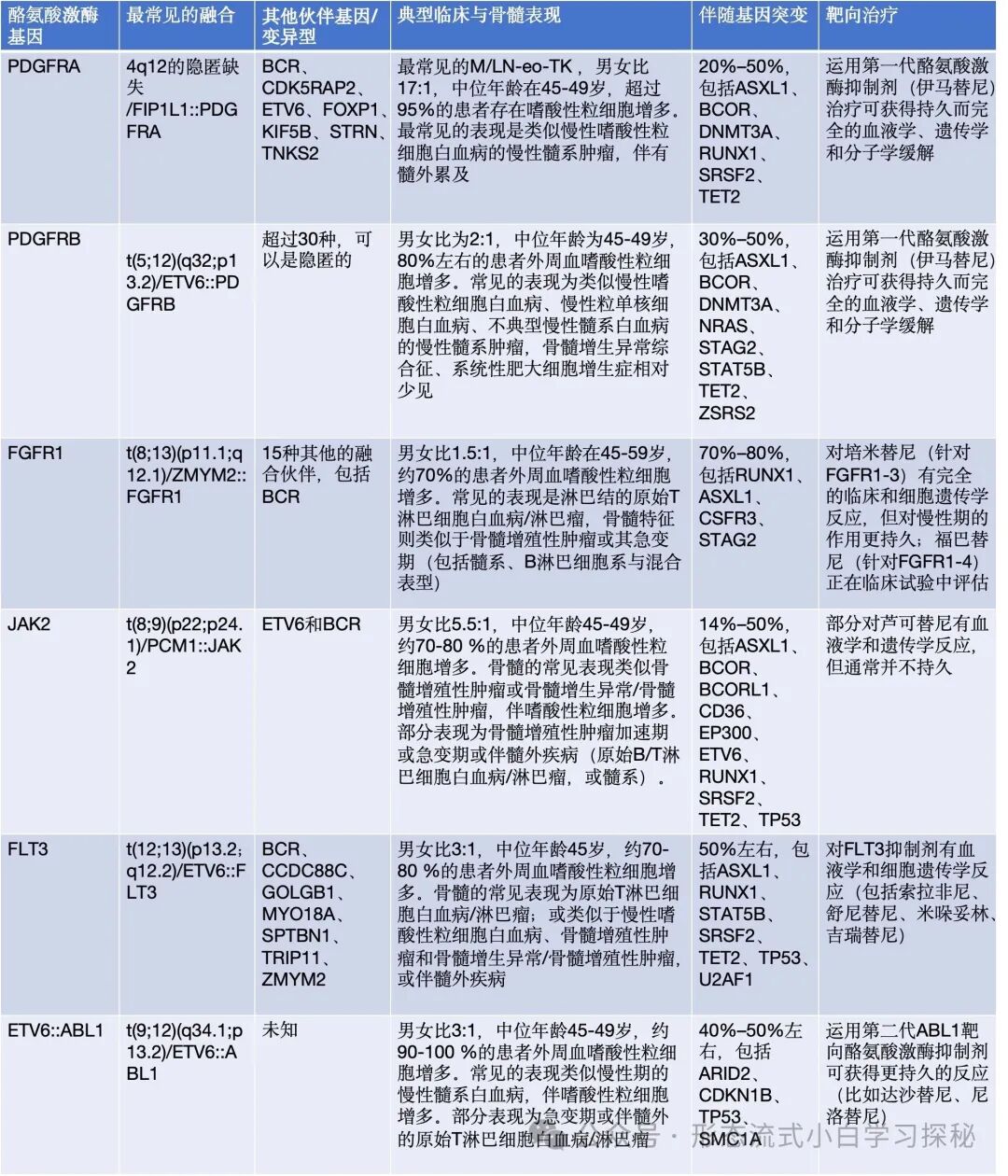
Peripheral blood eosinophilia is defined as a proportion of eosinophils in peripheral blood >6% and an absolute value >0.5×10^9/L
Prognosis and Clinical Commentary
Almost all patients with PDGFRA fusion genes are sensitive to low-dose (100 mg daily) imatinib therapy. Recent data show that some patients can maintain a treatment-free remission state after discontinuing imatinib; for relapsed patients, reinitiating imatinib therapy can rapidly achieve a second remission. Primary or secondary resistance is relatively rare, but when it occurs, it is associated with T674I or D842V mutations in the ATP-binding domain of the PDGFRA gene, and has a poor prognosis. Such patients show some degree of response to second or third-generation tyrosine kinase inhibitors (TKIs), but the response is not durable; therefore, it is strongly recommended to bridge to allogeneic hematopoietic stem cell transplantation (HSCT) as soon as possible.
2. Myeloid/lymphoid neoplasms with eosinophilia and PDGFRB rearrangements
Definition
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) stem cells with PDGFRB gene rearrangements leading to constitutive activation of tyrosine kinase (TK). Most cases present as chronic myeloid neoplasms (CMN), with some cases presenting as acute leukemia, usually (but not always) accompanied by eosinophilia.
Synonyms: None
Epidemiology
There is a slight male predominance among patients, with a male-to-female ratio of 2:1. The age of onset is quite broad, ranging from children to the elderly, with a median age in the 40s.
General Clinical Features
The disease primarily presents as chronic myeloid neoplasms (CMN) with eosinophilia, rarely presenting as acute myeloid leukemia (AML), B-cell acute lymphoblastic leukemia/lymphoma (B-ALL/LBL), or T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/LBL). In cases presenting as CMN, systemic symptoms and symptoms related to anemia are more common. Splenomegaly is observed in 40% to 50% of patients, while hepatomegaly is less common. End-organ damage related to eosinophilia may occur, leading to intermittent pain (in the back, head, chest, or abdomen), rashes, and respiratory symptoms. Serum tryptase levels are often elevated, and some patients may also have elevated lactate dehydrogenase (LDH) levels.
Morphology and Immunophenotype
Leukocytosis is common, seen in 60% to 70% of patients; peripheral blood eosinophilia is observed in approximately 70% to 80% of patients, with significant eosinophilia seen in about 50% of patients. Due to the high white blood cell count, over 30% of patients may have an absolute count of peripheral blood monocytes ≥1.0 × 10⁹/L, but only about 15% to 20% of patients show both relative monocytosis (≥10%) and absolute monocytosis. Anemia is common, but thrombocytopenia is less common. Similar to myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene rearrangements (M/LN-eo-TK PDGFRA), eosinophilic morphology may be abnormal in about half of cases (see Figure 6.2).
Bone marrow shows significantly active cellular proliferation, with increased eosinophils, granulocytes, and in some cases, monocytes (see Figure 6.2). Megakaryocyte numbers may be increased, decreased, or normal, with morphology showing no significant abnormalities, pathological hematopoiesis, or occasionally presenting large cell morphology typical of myeloproliferative neoplasms. Similar to myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene rearrangements, scattered spindle-shaped mast cells may be observed in the bone marrow, and these mast cells may show abnormal CD25 expression, which can be highlighted by immunohistochemical staining (such as CD117 and tryptase staining) (see Figure 6.2). In rare cases, mast cells may form tightly clustered aggregates resembling systemic mastocytosis (SM). Approximately one-third of cases may show significant bone marrow fibrosis (MF-2 or MF-3 grade). Based on the characteristics of peripheral blood and bone marrow, these chronic myeloid neoplasms (CMNs) may be described as resembling chronic eosinophilic leukemia, not otherwise specified (CEL, NOS); chronic monocytic leukemia with eosinophilia (CMML); myelodysplastic syndromes (MDS); atypical chronic myeloid leukemia (aCML); myelodysplastic/myeloproliferative neoplasms, not otherwise specified (MDS/MPN, NOS); myeloproliferative neoplasms, unclassifiable (MPN-U); or systemic mastocytosis (SM). In some cases, patients may present with acute leukemia, lymphoblastic leukemia, and/or extramedullary tumors (EMDs).
Genetics
The PDGFRB gene is located at the 5q32 chromosomal position, and to date, more than 30 partner genes have been found to fuse with it, with t(5;12)(q32;p13.2)/ETV6::PDGFRB fusion being the most common (approximately 50%), followed by CCDC88C::PDGFRB fusion, while other fusion types are mostly reported in individual cases. Previously, it was believed that myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo) with PDGFRB gene rearrangements should show abnormalities in the 5q31-33 region in appropriate routine chromosomal karyotype analysis. However, recent studies have shown that cryptic PDGFRB gene rearrangements are quite common and often occur with other partner genes besides ETV6, such as DIAPH1, BCR, AFAP1L1, SART3, and GBP1. Cryptic fusions may be due to complex chromosomal karyotypes, with small fragment deletions, inversions, or structural variations that are difficult to identify. Additionally, some fusion types may not even be detectable by PDGFRB break probe fluorescence in situ hybridization (FISH) techniques. Therefore, if both chromosomal karyotype and FISH results are negative, and there is a clinical suspicion of tyrosine kinase fusions, further genetic testing (such as RNA sequencing, optical genome mapping, etc.) should be performed. PDGFRB gene rearrangements with partner genes such as EBF1, SSBP2, TNIP1, ZEB2, and ATF7IP are almost exclusively detected in de novo BCR::ABL1-like B-cell acute lymphoblastic leukemia (B-ALL), and not in myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK); while rearrangements of other partner genes may appear in both diseases. Next-generation sequencing (NGS) results show mutations in ASXL1, TET2, BCOR, ETV6, STAG2, and RUNX1 in 30% to 50% of cases. However, the prognostic impact of these mutations is currently unclear.
Prognosis and Clinical Commentary
Similar to cases with PDGFRA gene rearrangements, myeloid/lymphoid neoplasms with eosinophilia and PDGFRB gene rearrangements show excellent responses to imatinib, achieving long-term complete hematologic and molecular remission. For cases in the acute phase of chronic myeloid neoplasms (CMN) or those progressing from initial chronic phase, in addition to imatinib therapy, induction chemotherapy and/or allogeneic hematopoietic stem cell transplantation are usually employed. However, studies have shown that even in patients with acute transformation or those with myeloid sarcomas, imatinib monotherapy can achieve durable complete hematologic and molecular remission.
3. Myeloid/lymphoid neoplasms with eosinophilia and FGFR1 rearrangements
Definition
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) stem cells with FGFR1 gene rearrangements, leading to heterogeneity and complexity in disease phenotype.
Synonyms: None
Epidemiology
Male patients slightly outnumber female patients, with a male-to-female ratio of approximately 1.5:1. The disease typically occurs in individuals in their 40s to 50s.
General Clinical Features
A significant proportion of cases (60%-70%) present in the acute phase as lymphoblastic lymphoma, characterized by either increased blast cells in the bone marrow or presenting as extramedullary tumors (EMD), or myeloid sarcomas involving lymph nodes, mediastinum, or skin tissues. Approximately one-third of patients may have both lymphoblastic lymphoma in lymph nodes and chronic myeloid neoplasms (CMN) in the bone marrow (see Figure 6.3). Only 30% to 40% of cases present in the chronic phase. Patients often exhibit systemic symptoms such as fever, weight loss, and night sweats. Splenomegaly is common. Eosinophilia varies (70% of patients have it); however, significant eosinophilia is only seen in one-third of patients.

Figure 6.3 shows common clinical manifestations and histopathological findings of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK) associated with t(8;13)(p11;q12)/ZMYM2::FGFR1 rearrangements. Lymph node biopsy (A) shows proliferation of blast cells, with significant eosinophilic infiltration; immunohistochemical staining shows that these blast cells are primarily T-lymphoblasts, confirmed by CD3 (C) and terminal deoxynucleotidyl transferase (TdT, D) staining. Scattered immature myeloid cells can be seen in the bone marrow, which can be displayed by myeloperoxidase (E) staining. The bone marrow shows characteristics of myeloproliferative neoplasms, with significant eosinophilia (B). (A and B: Hematoxylin-Eosin stain; C: CD3 stain; D: TdT stain; E: myeloperoxidase stain; A-E: magnification 40×).
Morphology and Immunophenotype
For cases presenting as chronic myeloid neoplasms (CMN), the characteristics in peripheral blood and bone marrow may resemble MPN-U, CEL, NOS, MDS/MPN, CMML, aCML, and MDS/MPN, NOS. For cases presenting as acute leukemia or extramedullary tumors (EMD), the blast cells may be B-lymphoblasts, myeloid blast cells, mixed phenotype blast cells, or T-lymphoblasts. Cases associated with t(8;13)(p11;q12)/ZMYM2::FGFR1 rearrangements often present as nodular (or extranodal) T-lymphoblastic lymphoma with scattered or perivascular myeloid blast cells, historically labeled as “biphenotypic lymphoma” (see Figure 6.3). Cases initially presenting in the chronic phase may subsequently progress to acute leukemia in the bone marrow or extramedullary sites. Conversely, patients initially presenting as acute leukemia or EMD may still exhibit “abnormal blood counts and bone marrow abnormalities” after achieving remission, with FGFR1 fusion signals detectable in mature hematopoietic cells. If there is an increase in blast cells or immature cell proliferation in the tissue, flow cytometric immunophenotyping analysis and/or immunohistochemical staining should be performed to determine the phenotypic characteristics of the blast cells. Scattered spindle-shaped mast cells (CD117 and tryptase positive) are often observed, and abnormal CD25 expression is noted, but rarely do they form aggregates resembling systemic mastocytosis (SM).
The disease phenotype may vary due to different partner genes. t(8;22)(p11.2;q11.2)/BCR::FGFR1 rearrangements often present as chronic myeloid neoplasms (CML) or B-cell acute lymphoblastic leukemia (B-ALL) in the lymphoblastic phase of M/LN-eo-TK, without significant eosinophilia, while about half of t(8;9)(p12;q33)/CEP110::FGFR1 rearrangement cases present with monocytosis and tonsillar enlargement, likely due to leukemic involvement, while FGFR10P1::FGFR1 rearrangements often present with erythrocytosis.
Genetics
The FGFR1 gene is located at 8p11.2 on chromosome 8. To date, at least 16 partner genes have been reported to fuse with FGFR1 (see Table 6.1). The most common partner gene is ZMYM2, located at 13q12, followed by the BCR gene located at 22q11. FGFR1 gene rearrangements are usually not cryptic cytogenetically; however, due to the small size of the 8p11 region, the reported range of fusion bands may extend from 8p11.2 to 8p21. To establish a diagnosis, it is crucial to confirm whether FGFR1 is involved using fluorescence in situ hybridization (FISH) or RNA sequencing (RNAseq) techniques. Targeted next-generation sequencing (NGS) studies show somatic mutations in 70% to 80% of cases, with approximately 70% to 80% of mutations involving the RUNX1 gene, which is associated with acute leukemia presentation or disease progression.
Prognosis and Clinical Commentary
Many cases progress rapidly, with an overall survival of less than 3 years. Tumors with FGFR1 gene rearrangements show no response to first-generation tyrosine kinase inhibitors (TKIs, such as imatinib), and only exhibit some efficacy to third-generation TKIs—ponatinib. Based on the high clinical and complete cytogenetic remission rates, the FGFR1-3 inhibitor pemigatinib was approved by the U.S. Food and Drug Administration (FDA) in August 2022 for the treatment of adult patients with myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK) who have previously received at least one treatment. A clinical trial using futibatinib (an anti-FGFR1-4 drug) is ongoing. Allogeneic hematopoietic stem cell transplantation (HSCT) is considered the best treatment option, and drugs like pemigatinib may be particularly useful as bridging therapy for transplant-eligible candidates.
4. Myeloid/lymphoid neoplasms with eosinophilia and PCM1::JAK2 and its genetic variants ETV6::JAK2 and BCR::JAK2 fusion genes
Definition
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) hematopoietic stem cells carrying the PCM1::JAK2 gene fusion. Other recognized partner genes include the ETV6::JAK2 fusion gene located at t(9;12)(p24.1;p13.2) and the BCR::JAK2 fusion gene located at t(9;22)(p24.1;q11.2). Individual cases have been reported with other partner genes, including RPN1/t(3;9)(q21;p24), NF-E2/t(9;12)(p24;q13), RUNX1/t(9;21)(p24·2;q22.1), and PEX14/t(1;9)(p36;p24.1), with clinical presentations resembling myelodysplastic syndromes (MDSs) or myeloproliferative neoplasms, such as primary myelofibrosis or polycythemia vera. Although these rare gene rearrangements may belong to the same category, the information currently available is very limited. In BCR::ABL1-like B-cell acute lymphoblastic leukemia (B-ALL), most reports have documented fusions of JAK2 with partner genes such as ATF7IP, EBF1, GOLGA5, HMBOX1, OFD1, PAX5, PPFIBP1, RFX3, SMU1, SNX29, SSBP2, STRN3, TERF2, TPR, USP25, ZBTB46, ZNF274, and ZNF340. It should be noted that the PCM1::JAK2 fusion gene may be acquired during disease progression or relapse of acute myeloid leukemia (AML), and this situation should not be considered as myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK). Some mature T-cell lymphoma cases, including anaplastic large cell lymphoma and mycosis fungoides, have been found to carry the PCM1::JAK2 fusion gene. If there is no myeloid involvement, these cases do not fall under the M/LN-eo-TK category.
Synonyms: None
Epidemiology
Patients with the PCM1::JAK2 fusion gene have a median age of 50 years at diagnosis, with a wide age range, and a predominance of male patients (male-to-female ratio of 5.5:1). Based on the limited number of cases, the other two gene variant types also exhibit similar demographic characteristics.
General Clinical Features
Approximately half of the patients present in the chronic phase, often manifesting as CEL, NOS, MPN-U, and MDS/MPN; while the other half present in the accelerated or blast phase, or exhibit extramedullary disease (EMD). The blast cells are usually of myeloid origin; some may be of B-lymphoblastic origin; while T-lymphoblastic blast cells are extremely rare. Organomegaly (hepatosplenomegaly) and/or lymphadenopathy are very common.
Morphology and Immunophenotype
Peripheral blood eosinophilia is common among patients with chronic myeloid neoplasms (CMNs), with varying degrees from mild to significant. The morphology of eosinophils varies, from no obvious abnormalities to abnormal forms, including those with dysplastic features as previously described. Histopathological examination of bone marrow in patients carrying the PCM1::JAK2 fusion gene often shows a characteristic “triad” (Figure 6.4), which includes cellular proliferation with eosinophilic infiltration, a large aggregation of immature erythroid precursors (proerythroblasts), and bone marrow fibrosis. The aggregation of these immature erythroid precursors is very prominent, referred to as “erythroid microtumors.” In some cases, erythroid hyperplasia and dysplasia of the erythroid, granulocyte, and/or megakaryocyte lineages may be observed. In contrast, the two gene variant types t(9;12)(p24.1;p13.2)/ETV6::JAK2 and t(9;22)(p24.1;q11.2)/BCR::JAK2 often do not exhibit the aforementioned characteristic “triad.” Cases with the BCR::JAK2 fusion gene may resemble chronic myeloid leukemia (CML) with eosinophilia.
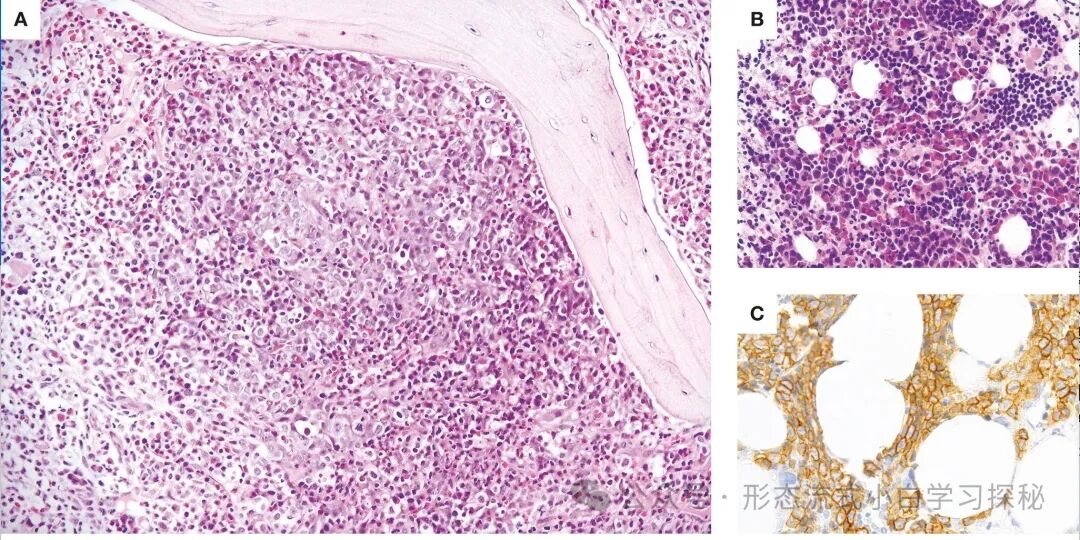
Figure 6.4 shows the characteristic manifestations of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK) with the PCM1::JAK2 fusion gene. Bone marrow cellular proliferation is evident, with a large aggregation of immature erythroid precursors (mainly proerythroblasts), eosinophilia, and often accompanied by bone marrow fibrosis (A). In some cases, immature erythroid precursors may exist in multiple large aggregates or sinusoidal aggregates (B), which can be highlighted by E-cadherin immunohistochemical staining (C). (A and B: Hematoxylin-Eosin stain; C: E-cadherin stain; A and B: magnification 20×; C: magnification 40×).
In extramedullary disease (EMD), eosinophilic infiltration and a large aggregation of immature erythroid precursors may be observed. If proerythroblast formation leads to extensive proliferation and disrupts the normal structure of extramedullary tissues, a diagnosis of erythroid myeloid sarcoma is appropriate. The presence of erythroid microtumors or extramedullary erythroid nodules can be confirmed by immunohistochemical staining with CD71, LMO2, and/or E-cadherin (Figure 6.4). For cases with increased blast cells, flow cytometric immunophenotyping analysis and immunohistochemical staining will help clarify the phenotypic characteristics of the blast cells.
Genetics
The JAK2 gene is located at the end of the short arm of chromosome 9. In the presence of sufficient mid-stage metaphases, the vast majority of PCM1::JAK2 fusion cases are not cryptic and will present t(8;9)(p22;p24.1) translocation. It is generally believed that in an appropriate clinical context, if t(8;9)(p22;p24) translocation is detected, it can be preliminarily inferred that PCM1::JAK2 fusion exists, but it is strongly recommended to perform fluorescence in situ hybridization (FISH) testing using probes targeting JAK2 for confirmation. The t(9;12)(p24.1;p13.2)/ETV6::JAK2 and t(9;22)(p24.1;q11.2)/BCR::JAK2 translocations are also usually not cryptic. For extramedullary disease (EMD), where chromosomal karyotype analysis is often not possible, if patients exhibit eosinophilia and erythroid microtumors, PCM1::JAK2 fusion should be highly suspected. Importantly, all these genetic abnormalities (PCM1::JAK2 and its variants) can be seen in BCR::ABL1-like B-cell acute lymphoblastic leukemia (B-ALL) and primary T-cell acute lymphoblastic leukemia (T-ALL); therefore, demonstrating the presence of a potential myeloid component is key when diagnosing myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK). In PCM1::JAK2-positive M/LN-eo-TK cases, mutations have been reported in 14% to 50% of cases, involving genes such as ASXL1, TET2, BCOR, RUNX1, SRSF2, ETV6, TP53, and EP300. However, mutation data for the t(9;12)(p24.1;p13.2)/ETV6::JAK2 and t(9;22)(p24.1;q11.2)/BCR::JAK2 translocations are relatively scarce.
Prognosis and Clinical Commentary
For patients presenting as chronic myeloid neoplasms (CMN), the disease may progress indolently, with a 5-year survival rate of up to 80%. On the other hand, for those diagnosed at the acute leukemia stage or whose condition progresses to acute leukemia, the prognosis is extremely poor. Targeted therapy using JAK2 inhibitors such as ruxolitinib may provide some but limited efficacy, and this treatment can be used as a bridging therapy for patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT), which may lead to a durable disease-free survival.
5. Myeloid/lymphoid neoplasms with eosinophilia and FLT3 rearrangements
Definition
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) stem cells with FLT3 gene rearrangements (leading to constitutive activation of tyrosine kinase [TK]). The most common fusion form is t(12;13)(p13;q12)/ETV6::FLT3, with approximately eight reported fusion partner genes.Synonyms: NoneEpidemiology
The disease appears to be more common in males, with a male-to-female ratio of approximately 2:1. The age of onset spans a wide range, with a median age in the 40s to 50s.
General Clinical Features
Patients typically present with peripheral blood abnormalities, including leukocytosis, monocytosis, and one or more cytopenias. Eosinophilia is common, occurring in about two-thirds of patients, but may be mild. Lymphadenopathy, splenomegaly, or extramedullary disease (EMD) manifestations are also common, seen in over 50% of cases. Clinical features may include fatigue, easy bruising, rashes, or symptoms related to extramedullary involvement.
Morphology and Immunophenotype
Peripheral blood often shows eosinophilia, which may or may not be accompanied by morphological abnormalities of eosinophils. Some cases may exhibit monocytosis, leading to a diagnosis of chronic myelomonocytic leukemia (CMML). Bone marrow examination often shows a chronic myeloid neoplasm (CMN), with manifestations that may resemble MPN, NOS, myelodysplastic syndromes/myeloproliferative neoplasms (such as CMML), atypical chronic myeloid leukemia (aCML), chronic eosinophilic leukemia, or systemic mastocytosis associated with myeloid neoplasms. Blast cell counts may be increased. Some cases present as acute leukemia (such as acute myeloid leukemia [AML], T-cell acute lymphoblastic leukemia [T-ALL], or mixed phenotype acute leukemia [MPAL], rarely B-cell acute lymphoblastic leukemia [B-ALL]) or extramedullary disease (EMD), such as T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/LBL), myeloid sarcoma, mixed phenotype acute leukemia, and occasionally mature T-cell lymphoma. It has been reported that mature T-cell lymphoma cases may also be associated with chronic myeloid neoplasms in the bone marrow. Flow cytometric immunophenotyping and/or immunohistochemical staining are required to clarify the phenotypic characteristics of the blast cells in cases with increased blast cells.
Genetics
The FLT3 gene is located in the q12 region of chromosome 13, with the most common partner gene being ETV6 located at 12p13.2, accounting for about half of reported cases. ETV6::FLT3/t(12;13)(p13;q12) is usually not cryptic. Other reported partner genes include ZMYM2/13q12, TRIP11/14q32, SPTBN1/2p16, GOLGB1/3q13, CCDC88C/14q32, MYO18A/17q12, and BCR/22q11. Additionally, a few cases have reported FLT3 rearrangements with unidentified partner genes at 3q27, 5q15, 5q35, 7q36, and 13q22. Some fusions are more cryptic, such as ZMYM2::FLT3, requiring FISH or RNA sequencing for identification. Approximately half of the cases have mutations involving ASXL1, PTPN11, RUNX1, SETBP1, SRSF2, STAT5B, TET2, TP53, and U2AF1. The prognostic impact of these mutations is currently unclear.
Prognosis and Clinical Commentary
If untreated, this disease often has an aggressive clinical course, with early progression. Monotherapy with FLT3 inhibitors such as sunitinib, midostaurin, sorafenib, or gilteritinib has anti-FLT3 activity, and some of these drugs have been reported to induce hematologic remission, regardless of whether complete cytogenetic remission is achieved. Some patients may achieve sustained remission or stable disease with monotherapy with FLT3 inhibitors, but others may have shorter remission periods. Resistance may occur due to acquired FLT3 N841K mutations in the activation loop of the tyrosine kinase (TK) domain. Nevertheless, FLT3 inhibitors still help bridge patients to subsequent allogeneic hematopoietic stem cell transplantation (HSCT).
6. Myeloid/lymphoid neoplasms with eosinophilia and ETV6::ABL1
Definition:
A myeloid and/or lymphoid neoplasm originating from multipotent (lymphoid/myeloid) stem cells with the ETV6::ABL1 fusion gene (leading to constitutive activation of tyrosine kinase [TK]). It has been reported that the ABL1 gene can fuse with other partner genes; however, most of these cases present as B-cell acute lymphoblastic leukemia (B-ALL) similar to BCR::ABL1 gene fusion or de novo T-cell acute lymphoblastic leukemia (T-ALL). Therefore, caution should be taken to avoid misdiagnosing these cases as myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK). However, a few cases with alternative partner genes (such as PRRC2B::ABL1) have been reported to present as chronic myeloid neoplasms or mixed phenotype acute leukemia with eosinophilia. Such cases can be appropriately classified under the ETV6::ABL1 gene variant type within myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK).
Synonyms: None
Epidemiology
This disease primarily occurs in adults, with a predominance of male patients (male-to-female ratio of 3:1). The median age of onset is in the fourth to fifth decades of life (i.e., 40 to 50 years). Pediatric cases are mostly de novo T-cell acute lymphoblastic leukemia (T-ALL) or B-cell acute lymphoblastic leukemia (B-ALL) similar to BCR::ABL1 gene fusion, rather than myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK).
General Clinical Features
Patients often present with leukocytosis, eosinophilia (in >90% of cases, many patients also have hyper-eosinophilia), neutrophilia, monocytosis, and some may also have basophilia (>1%). These cases are often diagnosed as myeloproliferative neoplasms (MPN) with significant eosinophilia, or chronic eosinophilic leukemia (not otherwise specified, CEL, NOS), and occasionally as acute phases of myeloproliferative neoplasms. Additionally, it has been reported that some patients may have both chronic myeloid neoplasms (CMN) in the bone marrow and T-lymphoblastic lymphoma (T-LBL) in lymph nodes. Most patients exhibit splenomegaly.
Morphology and Immunophenotype
Peripheral blood often shows significant eosinophilia, which may or may not be accompanied by the aforementioned abnormalities. Bone marrow usually shows significantly active cellular proliferation, accompanied by eosinophilia. Described cases resemble chronic myeloid leukemia (CML) with eosinophilia; they also include chronic eosinophilic leukemia (not otherwise specified, CEL, NOS), Philadelphia chromosome-negative myeloproliferative neoplasms (Ph-negative MPN), atypical chronic myeloid leukemia (aCML), myelodysplastic syndromes/myeloproliferative neoplasms (not otherwise specified, MDS/MPN, NOS), and primary thrombocythemia. Occasionally, an increase in spindle-shaped mast cells may be observed. Extramedullary disease (EMDs) is mostly reported as T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/LBL). For cases with increased blast cells, flow cytometry and immunohistochemical staining are required for phenotypic identification.
Genetics
The ABL1 gene is located in the q34.12 region of chromosome 9, while its partner gene ETV6 is located in the p13.2 region of chromosome 12. The generation of the ETV6::ABL1 fusion gene often results from translocations and inversions involving the ETV6 gene in the 9q34 region or the ABL1 gene in the 12p13.2 region, or complex rearrangements involving the insertion of the ETV6 gene into the 9q34 region or the ABL1 gene into the 12p13.2 region. This fusion gene is often cryptic, thus requiring fluorescence in situ hybridization (FISH) analysis with probes targeting the ETV6 and ABL1 gene breaks, RNA sequencing (RNAseq) techniques, or reverse transcription polymerase chain reaction (RT-PCR) for detection. Through alternative splicing, this fusion gene produces two fusion transcripts, type A (lacking the fifth exon of the ETV6 gene) and type B (containing the fifth exon), with type B being significantly more prevalent than type A. In rare cases, other partner genes (such as PRRC2B) have been reported to exhibit the disease characteristics of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase rearrangements (M/LN-eo-TK) and respond to tyrosine kinase inhibitor (TKI) therapy, which can be considered as variants of ETV6::ABL1. Additionally, mutations involving genes such as ARID2, TP53, SETD2, CDKN1B, PTPN11, and SMC1A have been detected in approximately 50% of cases.
Prognosis and Clinical Commentary
Most patients present in the chronic phase; however, in 30% of cases, the disease may progress to the acute phase or exhibit extramedullary disease (EMD), which may present as acute myeloid leukemia (AML), B-cell acute lymphoblastic leukemia (B-ALL), T-cell acute lymphoblastic leukemia (T-ALL), or myeloid sarcoma. Patients carrying the ETV6::ABL1 fusion gene show variable responses to targeted therapy with tyrosine kinase inhibitors (TKIs). In terms of induction treatment response, second-generation or third-generation tyrosine kinase inhibitors appear to be superior to imatinib. Among many patients presenting in the chronic phase, durable hematologic and molecular remissions have been observed, but this has not been seen in patients in the acute phase. For patients whose disease continues to progress despite the addition of tyrosine kinase inhibitors to standard chemotherapy, the prognosis is usually poor.