Definition:Chronic Myeloid Leukemia (CML) is caused by the chromosomal translocation (9;22)(q34.1;q11.2), which leads to the formation of the Philadelphia (Ph) chromosome containing the BCR::ABL1 fusion gene. The BCR::ABL1 oncogenic protein acts as a constitutively activated tyrosine kinase, driving malignant transformation by activating multiple signaling pathways. Granulocytes are the main proliferative component, but all myeloid lineages as well as some lymphoid and endothelial cells carry the abnormal gene. The natural course of untreated CML progresses in a biphasic or triphasic manner: the chronic phase (CML-CP) can progress to the accelerated phase (CML-AP) and blast phase (CML-BP). It is important to note that the blast phase can progress directly from CML-CP without passing through the accelerated phase.CML Diagnostic Criteria: The Ph chromosome or BCR::ABL1 fusion gene must be detected in the clinical and laboratory context. Patients need to undergo complete blood count and leukocyte differential, as well as bone marrow examination, to obtain samples for complete chromosomal karyotype analysis and morphological assessment to clarify disease staging. Bone marrow biopsy is particularly valuable for patients suspected of disease progression and always requires assessment of the presence and degree of bone marrow fibrosis. Karyotype analysis of bone marrow cells helps confirm the diagnosis of CML.At least 20 metaphase spreads need to be analyzed. Over 95% of CML patients carry the Ph chromosome, but in about 3% to 5% of patients, the Ph chromosome is part of a “complex translocation” involving other chromosomes. In rare cases, “cryptic Ph chromosomes” that cannot be identified by classical karyotype analysis may occur, requiring molecular detection for diagnosis. Additionally, performing quantitative reverse transcription polymerase chain reaction to baseline measure BCR::ABL1 transcript levels is necessary for disease monitoring.EpidemiologyThe annual incidence of Chronic Myeloid Leukemia (CML) is 1.0 to 1.5 per 100,000, with no significant racial or geographic differences, accounting for approximately 15% of newly diagnosed adult leukemia cases. Patients receiving modern treatment with BCR::ABL1 tyrosine kinase inhibitors have a life expectancy close to that of age-matched healthy individuals. Therefore, the prevalence of CML continues to rise and is expected to become the most common myeloid tumor. The only clearly defined risk factor for CML is exposure to ionizing radiation.General Clinical FeaturesCML patients may present withsystemic symptoms, including fatigue, weight loss, and night sweats. Splenomegaly is common, and hepatomegaly may also occur (reflecting extramedullary hematopoiesis). Significant lymphadenopathy is rare; if present, lymph node biopsy should be considered to rule out possible blast phase. The median age at diagnosis is 65 years, but CML can occur at any age, including in children. Males are slightly more affected than females. Over 50% of newly diagnosed patients are asymptomatic at diagnosis, often diagnosed due to persistent unexplained leukocytosis. The characteristic Ph chromosome t(9;22)(q34;q11) can be identified through routine cytogenetic examination or by fluorescence in situ hybridization or molecular studies detecting BCR::ABL1 abnormalities. Most patients are diagnosed during the chronic phase of Chronic Myeloid Leukemia (CML-CP), but about 5% of patients initially present as CML-AP or CML-BP (without a clear chronic phase history). Patients need to undergo bone marrow examination to obtain samples for karyotype and morphological assessment to clarify disease staging.If effective treatment is not administered, patients will show a trend of persistent deterioration in hematological parameters. Disease progression is usually associated with additional genetic abnormalities. Recognizing disease progression is important, but the clinical and morphological boundaries between CML-CP, CML-AP, and CML-BP are not clearly defined, and the parameters used to define these stages vary across different guidelines. However, recent studies emphasize that defining the progression from CML-CP to CML-AP is crucial for assessing TKI treatment response, selecting novel therapies, and making decisions regarding allogeneic hematopoietic stem cell transplantation.MorphologyCML-CPThe peripheral blood of patients with Chronic Myeloid Leukemia chronic phase (CML-CP) typically shows neutrophilia (Figure 1.1), with most cells at the mature neutrophil and myelocyte stages (the latter referred to as the “myelocyte peak”). Cell function is generally normal, and patients with CML-CP do not have an increased risk of infection or bleeding (Table 1.1). There are no significant developmental abnormalities, and blast cells usually account for less than 2% of white blood cells. Absolute basophilia is a typical finding, which may be accompanied by mild eosinophilia (significant elevation requires differentiation from other diseases). Absolute monocyte counts may be elevated (>1×10⁹/L), but the proportion of monocytes is usually less than 3%. Anemia is common, but it is relatively rare for hemoglobin levels to be below 10 g/dL at initial diagnosis. Platelet counts may be mildly or moderately elevated, but in about 10% of cases, platelet counts are significantly elevated (>1000×10⁹/L), requiring differentiation from other myeloproliferative neoplasms (such as essential thrombocythemia). In these cases, the white blood cell count may be normal or only mildly elevated, mimicking other subtypes of myeloproliferative neoplasms, such as essential thrombocythemia. Additionally, leukocytosis composed almost entirely of segmented neutrophils may resemble chronic neutrophilic leukemia (CNL), while cases with elevated absolute monocyte counts may resemble chronic monocytic leukemia (CMML).This atypical presentation of CML is often related to variations in the size of the BCR::ABL1 fusion protein caused by differences in the BCR gene breakpoint.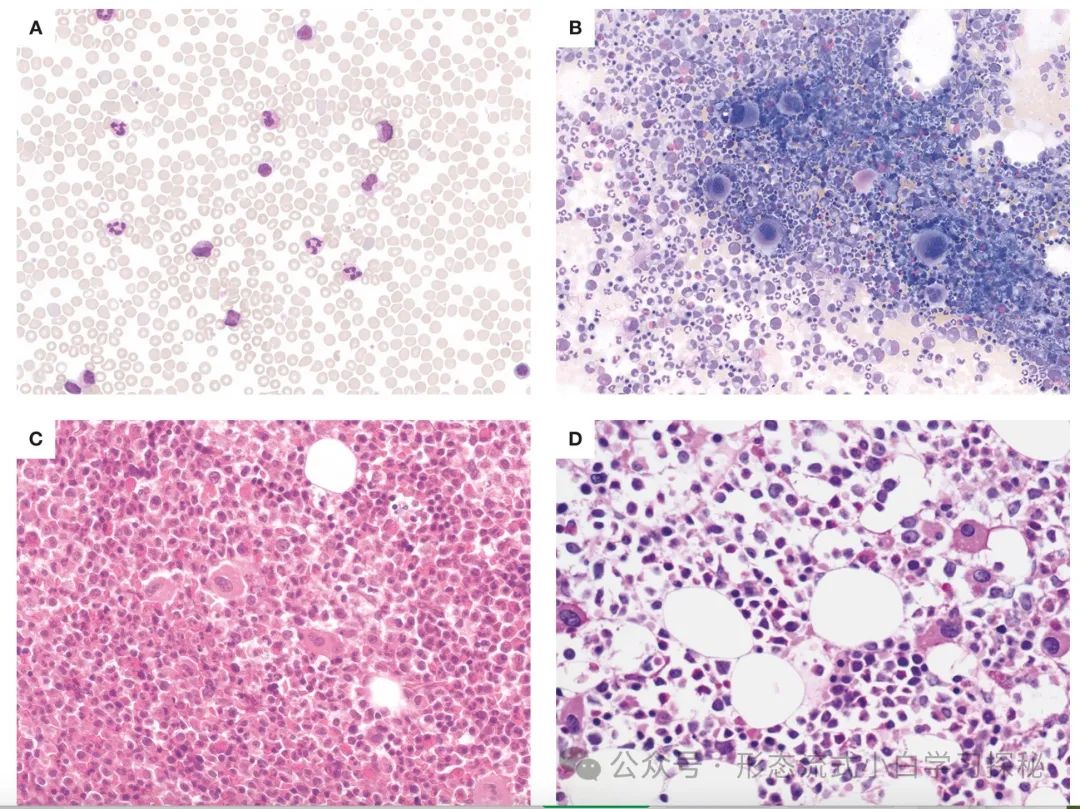 Figure 1.1 Chronic Myeloid Leukemia Chronic Phase (CML-CP)A. Peripheral blood smear shows significant leukocytosis, with a predominance of neutrophil differentiation lineages, mainly myelocytes and segmented neutrophils;B. Bone marrow smear shows significant granulocyte proliferation, with increased megakaryocytes (often with poorly lobulated nuclei, referred to as “dwarf megakaryocytes”);C. Bone marrow biopsy sections show a significant increase in granulocyte to erythroid ratio, with increased eosinophils and small megakaryocytes with reduced nuclear lobulation;D. CML-CP with p190-BCR::ABL1 subtype: small, round, atypical dwarf megakaryocytes are seen, with foamy/striped cytoplasm in the background.(Staining methods: A, B are Wright-Giemsa stain; C, D are hematoxylin-eosin stain; magnification: A, B are 60×; C, D are 40×)Table 1.1 Diagnostic features of chronic phase chronic myeloid leukemia
Figure 1.1 Chronic Myeloid Leukemia Chronic Phase (CML-CP)A. Peripheral blood smear shows significant leukocytosis, with a predominance of neutrophil differentiation lineages, mainly myelocytes and segmented neutrophils;B. Bone marrow smear shows significant granulocyte proliferation, with increased megakaryocytes (often with poorly lobulated nuclei, referred to as “dwarf megakaryocytes”);C. Bone marrow biopsy sections show a significant increase in granulocyte to erythroid ratio, with increased eosinophils and small megakaryocytes with reduced nuclear lobulation;D. CML-CP with p190-BCR::ABL1 subtype: small, round, atypical dwarf megakaryocytes are seen, with foamy/striped cytoplasm in the background.(Staining methods: A, B are Wright-Giemsa stain; C, D are hematoxylin-eosin stain; magnification: A, B are 60×; C, D are 40×)Table 1.1 Diagnostic features of chronic phase chronic myeloid leukemia In the chronic phase of Chronic Myeloid Leukemia (CML-CP), the bone marrow is almost always hypercellular (Figure 1.1), with significant granulocyte proliferation and expansion of myelocytes and segmented neutrophils. There are no significant developmental abnormalities. Blast cells usually account for less than 5% of bone marrow cells, while 10% or more of immature cells often indicate disease progression to CML accelerated phase (Table 1.2). Myelocyte expansion is a typical finding, often accompanied by eosinophilia/basophilia and reduced erythroid precursors. Commonly seen are Gaucher-like cells or “sea-blue” histiocytes (Figure 1.1), associated with increased cellular load and hypermetabolism. The number of megakaryocytes may be normal or reduced, but about 50% of cases show moderate to significant megakaryocyte proliferation.In most cases, the majority are dwarf megakaryocytes (small size, reduced nuclear lobulation), which need to be differentiated from small megakaryocytes in myelodysplastic syndromes (MDS). If a large number of MDS-like small megakaryocytes are found, caution should be exercised regarding the risk of disease progression.
In the chronic phase of Chronic Myeloid Leukemia (CML-CP), the bone marrow is almost always hypercellular (Figure 1.1), with significant granulocyte proliferation and expansion of myelocytes and segmented neutrophils. There are no significant developmental abnormalities. Blast cells usually account for less than 5% of bone marrow cells, while 10% or more of immature cells often indicate disease progression to CML accelerated phase (Table 1.2). Myelocyte expansion is a typical finding, often accompanied by eosinophilia/basophilia and reduced erythroid precursors. Commonly seen are Gaucher-like cells or “sea-blue” histiocytes (Figure 1.1), associated with increased cellular load and hypermetabolism. The number of megakaryocytes may be normal or reduced, but about 50% of cases show moderate to significant megakaryocyte proliferation.In most cases, the majority are dwarf megakaryocytes (small size, reduced nuclear lobulation), which need to be differentiated from small megakaryocytes in myelodysplastic syndromes (MDS). If a large number of MDS-like small megakaryocytes are found, caution should be exercised regarding the risk of disease progression.
Table 1.2 Diagnostic criteria for accelerated phase and blast phase chronic myeloid leukemia

a Second Philadelphia chromosome, trisomy 8, isochromosome 17q, trisomy 19, complex karyotype ≥3 cytogenetic abnormalities, or abnormalities of 3q26.2.
b Extramedullary blast proliferation, for lesions showing a diffuse (non–mass forming) blast proliferation.
c Immunophenotypic analysis is required to confirm lymphoid lineage.

a. Second Philadelphia chromosome, +8, long arm of chromosome 17 (i(17q)), +19, complex karyotype (≥3 cytogenetic abnormalities) or 3q26.2 abnormalities;
b. Extramedullary blast proliferation (referring to non-mass forming diffuse blast infiltration);c. Immunophenotypic analysis (such as flow cytometry) is required to confirm lymphoid differentiation.In the chronic phase of Chronic Myeloid Leukemia (CML-CP), bone marrow biopsy sections (Figure 1.1) show an increased number of immature granulocytes, which are usually arranged in layers along the trabeculae (typically 5-10 cells thick), gradually differentiating into mature granulocytes towards the central trabecular space. A small number of blast cells may be scattered near the trabeculae and/or in mature cells, but clusters of immature cells do not occur in CML-CP, although myeloid cell aggregates may be observed. Approximately 30%-40% of cases may show mild (MF-1) to significant (MF-2) reticulin fibrosis at diagnosis, which may be accompanied by increased megakaryocytes and splenomegaly (Figure 1.2).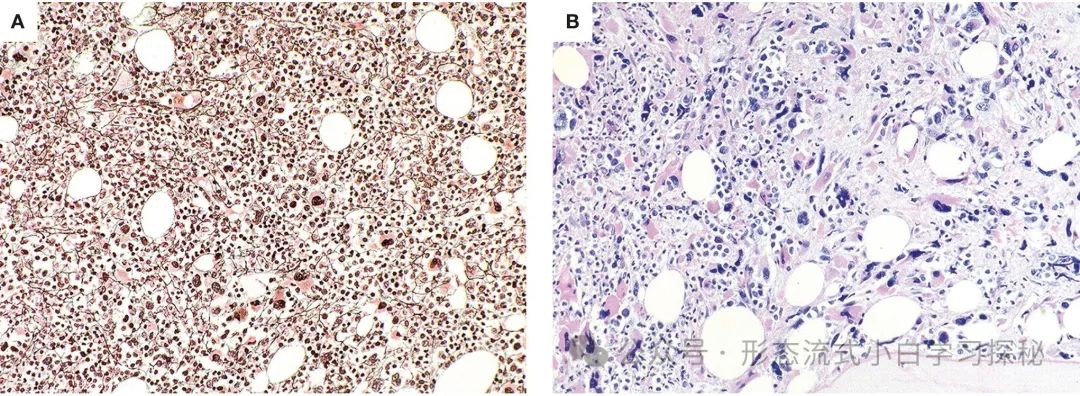 Figure 1.2 Chronic Myeloid Leukemia Chronic Phase (CML-CP) A. Bone marrow biopsy shows hypercellularity, with mild reticulin fibrosis (MF-1 grade) and increased small atypical megakaryocytes; B. Significant bone marrow fibrosis (MF-3 grade) at initial diagnosis, mimicking other myeloproliferative neoplasm subtypes, with significant atypical small megakaryocytes visible. (Staining methods: A, B are hematoxylin-eosin stain; magnification: A, B are 40×)Accelerated PhaseAlthough the diagnosis of CML primarily relies on BCR::ABL1 rearrangement detection, the clinical and morphological boundaries between CML-CP (chronic phase) and CML-AP (accelerated phase) are not clearly defined, and different guidelines have varying parameters for defining the transitional phase. Furthermore, CML-AP and CML-BP (blast phase) share similar gene expression profiles, with expression changes potentially occurring before clinical or morphological evidence of progression in late CML-CP or early CML-AP. However, as emphasized by recent data, accurately detecting disease progression remains important, as patients progressing from CML-CP to CML-AP generally have a worse prognosis, even in the era of modern TKI therapy. These patients need to be identified using easily applicable standards to include them in clinical trials of novel therapies (including assessments for allogeneic hematopoietic stem cell transplantation). Additionally, among patients receiving TKI therapy, 20% to 30% may experience significant disease progression due to acquired mutations affecting the tyrosine kinase binding domain of BCR::ABL1, rendering TKI ineffective. Less commonly, leukemic subclones with BCR::ABL1 amplification or SH3-SH2 domain mutations may lead to disease progression, or there may be genetic changes occurring in BCR::ABL1-independent signaling pathways.
Figure 1.2 Chronic Myeloid Leukemia Chronic Phase (CML-CP) A. Bone marrow biopsy shows hypercellularity, with mild reticulin fibrosis (MF-1 grade) and increased small atypical megakaryocytes; B. Significant bone marrow fibrosis (MF-3 grade) at initial diagnosis, mimicking other myeloproliferative neoplasm subtypes, with significant atypical small megakaryocytes visible. (Staining methods: A, B are hematoxylin-eosin stain; magnification: A, B are 40×)Accelerated PhaseAlthough the diagnosis of CML primarily relies on BCR::ABL1 rearrangement detection, the clinical and morphological boundaries between CML-CP (chronic phase) and CML-AP (accelerated phase) are not clearly defined, and different guidelines have varying parameters for defining the transitional phase. Furthermore, CML-AP and CML-BP (blast phase) share similar gene expression profiles, with expression changes potentially occurring before clinical or morphological evidence of progression in late CML-CP or early CML-AP. However, as emphasized by recent data, accurately detecting disease progression remains important, as patients progressing from CML-CP to CML-AP generally have a worse prognosis, even in the era of modern TKI therapy. These patients need to be identified using easily applicable standards to include them in clinical trials of novel therapies (including assessments for allogeneic hematopoietic stem cell transplantation). Additionally, among patients receiving TKI therapy, 20% to 30% may experience significant disease progression due to acquired mutations affecting the tyrosine kinase binding domain of BCR::ABL1, rendering TKI ineffective. Less commonly, leukemic subclones with BCR::ABL1 amplification or SH3-SH2 domain mutations may lead to disease progression, or there may be genetic changes occurring in BCR::ABL1-independent signaling pathways.
CML-AP (Table 1.2) is defined as having 10% to 19% blast cells in bone marrow or peripheral blood, peripheral blood basophils ≥20%, or the presence of additional clonal cytogenetic abnormalities in Ph+ cells, such as a second Ph chromosome, trisomy 8, long arm of chromosome 17, trisomy 19, complex karyotype, or 3q26.2 abnormalities. Previously used criteria for defining accelerated phase, such as treatment-independent thrombocytopenia (≤100×10⁹/L) or treatment-resistant thrombocytosis (>1000×10⁹/L) and splenomegaly, have been abolished. Additionally, the definition of CML-AP has excluded cases that do not achieve a complete hematological response, are resistant to sequential TKI therapy, or have >2 mutations of BCR::ABL1 during treatment.
The degree of hypercellularity in bone marrow samples from CML-AP patients can vary, with granulocytes and other myeloid lineages potentially showing developmental abnormalities. In smears or sections, myeloid blast cells (10% to 19%) may increase, and immunohistochemical staining for CD34 can be used to highlight them (Figure 1.3). Large clusters of megakaryocytes may sometimes be seen, including true small megakaryocytes resembling myelodysplastic syndromes, often accompanied by significant reticulin or collagen fibrosis. The presence of lymphoblasts in blood or bone marrow (confirmed by flow cytometry) should raise suspicion for impending lymphoblastic crisis (Figure 1.3), as lymphoblastic CML-BP has been reported to sometimes present acutely, usually without prior signs of CML-AP.
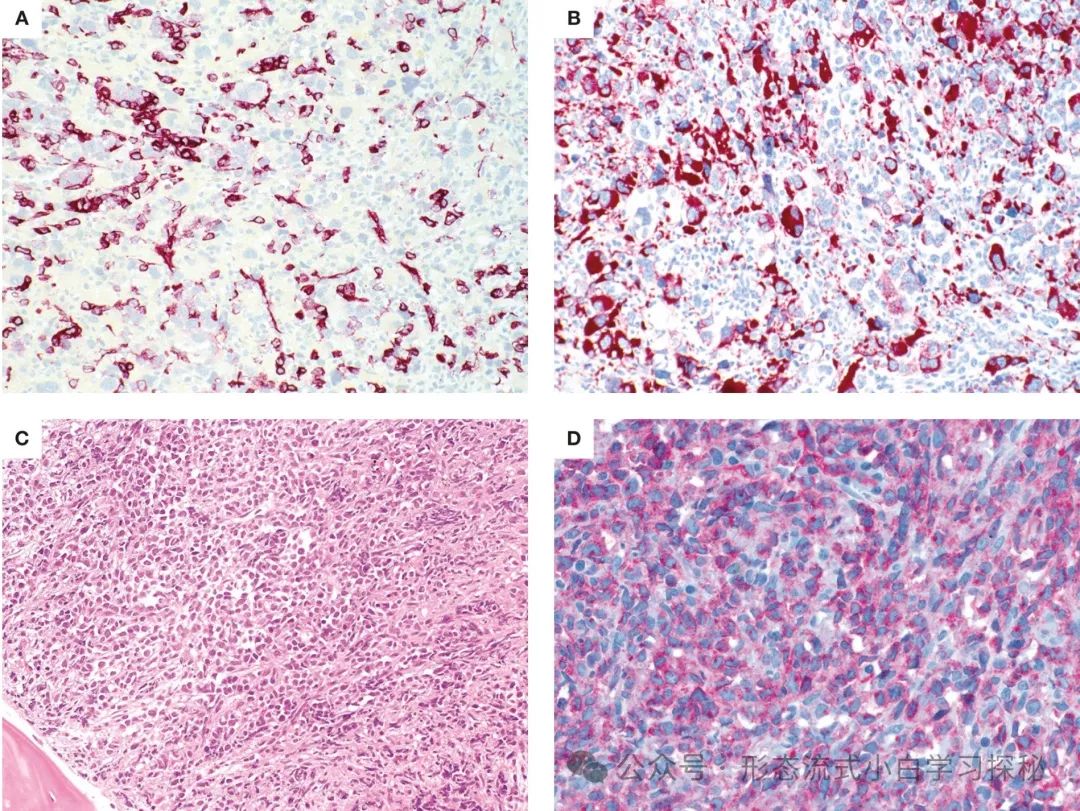
Figure 1.3 Chronic Myeloid Leukemia Accelerated Phase and Blast Phase (CML-AP/CML-BP) A. CML-AP bone marrow biopsy shows increased CD34+ blast cells (CD34 immunohistochemical staining); B. CML-AP with significant atypical megakaryocytes (CD61 marked), showing myelodysplastic-like morphology; C. Lymphoblastic crisis: blast cell proliferation, morphology showing lymphoid/lymphoblastic characteristics (HE staining); D. Lymphoblasts express CD19 (CD19 immunohistochemical staining). (Staining methods and magnification: A-D are all 40×)
Blast Phase
CML-BP is characterized by ≥20% blast cells in bone marrow or peripheral blood, or extramedullary blast proliferation destroying the structure of the affected tissue, i.e., extramedullary myeloid sarcoma. The presence of ≥5% lymphoblasts in peripheral blood or bone marrow defines lymphoblastic crisis, which requires further laboratory and genetic examination. It should be noted that other classification and risk stratification systems (such as the International Bone Marrow Transplant Registry, MD Anderson Cancer Center, European LeukemiaNet) define the threshold for blast cells in CML-BP as >30%, which is often used as eligibility criteria for clinical trials. Furthermore, even if the remaining bone marrow regions still show chronic phase (CML-CP) characteristics, if a single trabecular interspace is completely occupied by blast cells, a diagnosis of CML-BP can still be inferred.In most cases of CML-BP, the blast cells belong to the myeloid lineage (neutrophils, monocytes, megakaryocytes, basophils, eosinophils, erythroid blast cells, or their mixtures); however, 20% to 30% of cases consist of lymphoblasts, usually of B-cell origin (Figure 1.3), although cases of T-lymphoblastic and NK-cell transformation have also been reported. Sequential transformations may occur (e.g., myeloid transformation following lymphoblastic transformation). The source of blast cells is usually evident morphologically, but sometimes the blast cells may be primitive or heterogeneous, with mixed phenotypes being common; therefore, it is recommended to use multiparameter flow cytometry for immunophenotypic analysis. In rare cases, myeloid CML-BP cases may exhibit cytogenetic abnormalities, such as specific recurrent chromosomal translocations associated with acute myeloid leukemia (e.g., inv(16)(p13.1q22) or t(16;16)(p13.2;q22)), which appear in the same cell as the Ph chromosome. However, in such cases, these chromosomal abnormalities, which are usually associated with favorable prognosis in acute leukemia, do not have this effect when combined with the Ph chromosome.Extramedullary TissueIn CML-CP, the spleen may enlarge to varying degrees due to granulocyte infiltration of the red pulp. Similar infiltration may be seen in liver sinusoids and occasionally in lymph nodes. These extramedullary infiltrations consist of a mixture of immature and mature granulocytes; any significant shift towards immaturity with a blast cell percentage ≥10% suggests disease progression or transformation. In CML-BP, any extramedullary tissue may be infiltrated, but the most common sites are the spleen, liver, lymph nodes, skin, and soft tissues. Any clinical evidence of infiltrative lesions in any extramedullary site, regardless of blood and bone marrow examination results, should be investigated to rule out CML-BP.
Immunophenotyping
In CML-CP, the diagnostic value of immunophenotypic analysis is limited. However, some data suggest that abnormal expression of CD7, CD56, or CD11b on CD34+ stem cells may affect response to TKI therapy. However, the primary role of flow cytometry is to perform phenotypic analysis in CML-BP, including detecting mixed phenotypes. If the number of blast cells in the blood is insufficient and a bone marrow aspirate sample cannot be obtained, immunohistochemical staining (such as CD34, CD117) can assist in diagnosis.
In myeloid blast cell stages, blast cells may exhibit strong, weak, or no myeloperoxidase activity but will express antigens associated with granulocyte, monocyte, megakaryocyte, or erythroid differentiation, such as CD33, CD13, CD14, CD11c, CD11b, CD117, CD15, CD41, CD61, CD71, or blood group glycoproteins A or C. However, in many myeloid-derived cases, blast cells may also express one or more lymphocyte-associated antigens. Most lymphoblastic cases originate from B-cell precursors, expressing B-cell-related antigens (CD19, CD10, CD79a, PAX5, CD20) as well as terminal deoxynucleotidyl transferase, but a few cases express T-cell-related antigens (CD3, CD2, CD5, CD4, CD8, CD7). In both B-cell and T-cell derived blast cell stages, it is common to express one or more myeloid-related antigens. In the era of TKIs, the incidence of unusual immunophenotypes and blast cell types (e.g., basophilic blast cells, blast megakaryocytes) has increased.
Genetics
CML is characterized by the chromosomal translocation (9;22)(q34;q11), forming the Philadelphia chromosome (Ph) and generating the BCR::ABL1 fusion gene, which encodes a fusion protein with constitutively activated tyrosine kinase activity. The Philadelphia chromosome is not unique to CML, being found in 15% to 30% of adult and 5% of pediatric B-lymphoblastic leukemia (B-ALL), some mixed phenotype acute leukemias (MPAL), and rare acute myeloid leukemias (AML). The fusion protein encoded by the BCR::ABL1 fusion gene has abnormal tyrosine kinase activity, driving abnormal cell proliferation by activating downstream signaling pathways (such as JAK/STAT, PI3K/AKT, RAS/MEK, mTOR, and Src kinases).Different breakpoints and rearrangements determine the disease phenotype: in 95% of Chronic Myeloid Leukemia (CML) patients and 25% to 30% of Ph-positive acute lymphoblastic leukemia (Ph+ ALL) cases, the breakpoint is located in the major breakpoint cluster region of the BCR gene (spanning exons 12 to 16), forming the abnormal p210 fusion protein. In rare cases, the breakpoint is located in the μ breakpoint region of the BCR gene (spanning exons 17 to 20), generating a larger p230 fusion protein. These patients typically present with significant neutrophilia or thrombocytosis in peripheral blood; in about 1% to 2% of CML patients, the BCR gene’s minor breakpoint region (exons 1-2) breaks to generate the short p190 fusion protein (commonly seen in Ph-positive acute lymphoblastic leukemia, i.e., Ph+ ALL), with most patients detectable for trace p190 transcripts due to selective splicing of the BCR gene. CML patients carrying p190 may present with monocytosis, resembling chronic monocytic leukemia (CMML). In patients with CML-CP who are resistant to first or second-generation TKIs, about 30% have BCR::ABL1 kinase domain mutations. More than 100 mutations have been identified, with the T315I mutation being the most common (approximately 13%-16%), E255K and M351T mutations accounting for 9% to 14%, and mutations such as Y253F, Q252H, F317L accounting for 3% to 6%, while the V299L mutation is relatively rare. Compound mutations (≥2 mutations present on the same BCR::ABL1 allele, which may occur sequentially or simultaneously) exhibit different resistance profiles compared to single mutations. BCR::ABL1-independent TKI resistance mechanisms include stem cells activating alternative signaling pathways (such as Src, JAK/STAT, RAS/MAPK, and PI3K/AKT), genomic instability, epigenetic dysfunction, and immune system dysfunction. The activation of alternative pathways may explain why disease can still relapse in patients with good treatment responses.
In patients with CML-CP who are resistant to first or second-generation TKIs, about 30% have BCR::ABL1 kinase domain mutations. More than 100 mutations have been identified, with the T315I mutation being the most common (approximately 13%-16%), E255K and M351T mutations accounting for 9% to 14%, and mutations such as Y253F, Q252H, F317L accounting for 3% to 6%, while the V299L mutation is relatively rare. Compound mutations (≥2 mutations present on the same BCR::ABL1 allele, which may occur sequentially or simultaneously) exhibit different resistance profiles compared to single mutations. BCR::ABL1-independent TKI resistance mechanisms include stem cells activating alternative signaling pathways (such as Src, JAK/STAT, RAS/MAPK, and PI3K/AKT), genomic instability, epigenetic dysfunction, and immune system dysfunction. The activation of alternative pathways may explain why disease can still relapse in patients with good treatment responses.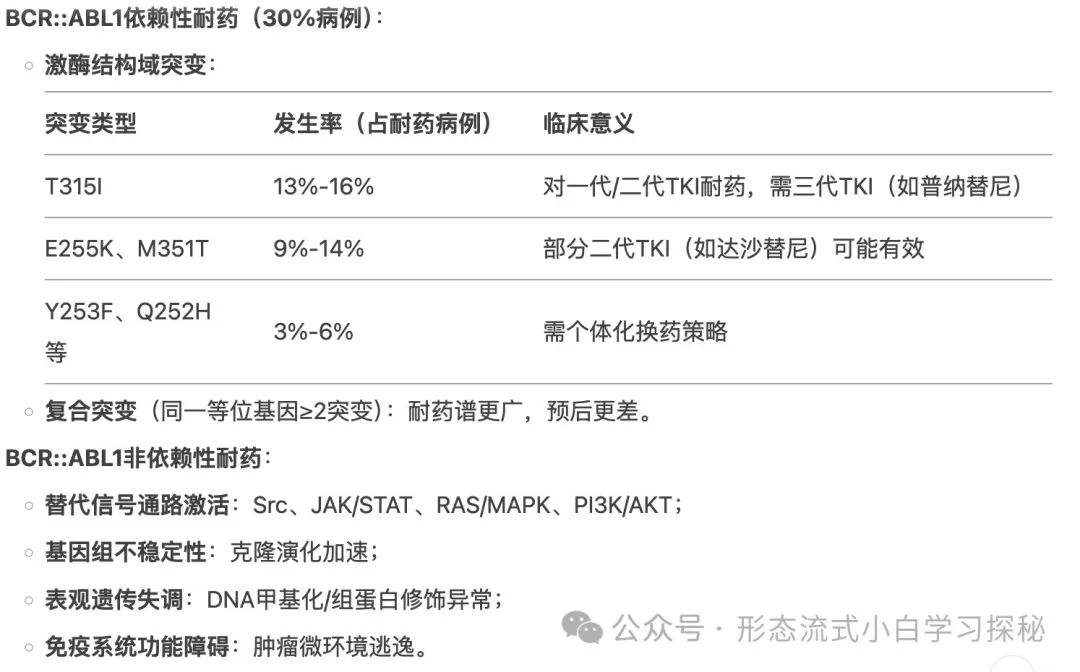 During the progression of CML-AP and CML-BP, secondary chromosomal abnormalities can be detected, with the most common being +8 (34% of cases with additional changes), +Ph (30%), i(17q) (20%), +19 (13%), -Y (8% in males), +21 (7%), +17 (5%), and monosomy 7 (5%), which are usually associated with poor prognosis. Additional chromosomal abnormalities acquired during treatment are considered markers of disease progression, with higher relapse rates in second-line or subsequent therapies. At diagnosis, 30% to 40% of Ph+ clones may exhibit gene mutations other than BCR::ABL1, with the most common being epigenetic regulatory genes such as ASXL1, DNMT3A, EZH2, IDH1, IDH2, and TET2. While ASXL1 mutations may occur in CML-CP, as the disease progresses to CML-BP, patients acquire additional mutations, including RUNX1, TP53, BCOR, and SETD1B. In rare cases, mutations coexisting in both Ph+ and Ph- clones suggest the presence of pre-existing mutations before acquiring BCR::ABL1. In high-risk CML patients, next-generation sequencing can highly sensitively identify emerging resistance mutations and should be incorporated into clinical decision-making.
During the progression of CML-AP and CML-BP, secondary chromosomal abnormalities can be detected, with the most common being +8 (34% of cases with additional changes), +Ph (30%), i(17q) (20%), +19 (13%), -Y (8% in males), +21 (7%), +17 (5%), and monosomy 7 (5%), which are usually associated with poor prognosis. Additional chromosomal abnormalities acquired during treatment are considered markers of disease progression, with higher relapse rates in second-line or subsequent therapies. At diagnosis, 30% to 40% of Ph+ clones may exhibit gene mutations other than BCR::ABL1, with the most common being epigenetic regulatory genes such as ASXL1, DNMT3A, EZH2, IDH1, IDH2, and TET2. While ASXL1 mutations may occur in CML-CP, as the disease progresses to CML-BP, patients acquire additional mutations, including RUNX1, TP53, BCOR, and SETD1B. In rare cases, mutations coexisting in both Ph+ and Ph- clones suggest the presence of pre-existing mutations before acquiring BCR::ABL1. In high-risk CML patients, next-generation sequencing can highly sensitively identify emerging resistance mutations and should be incorporated into clinical decision-making.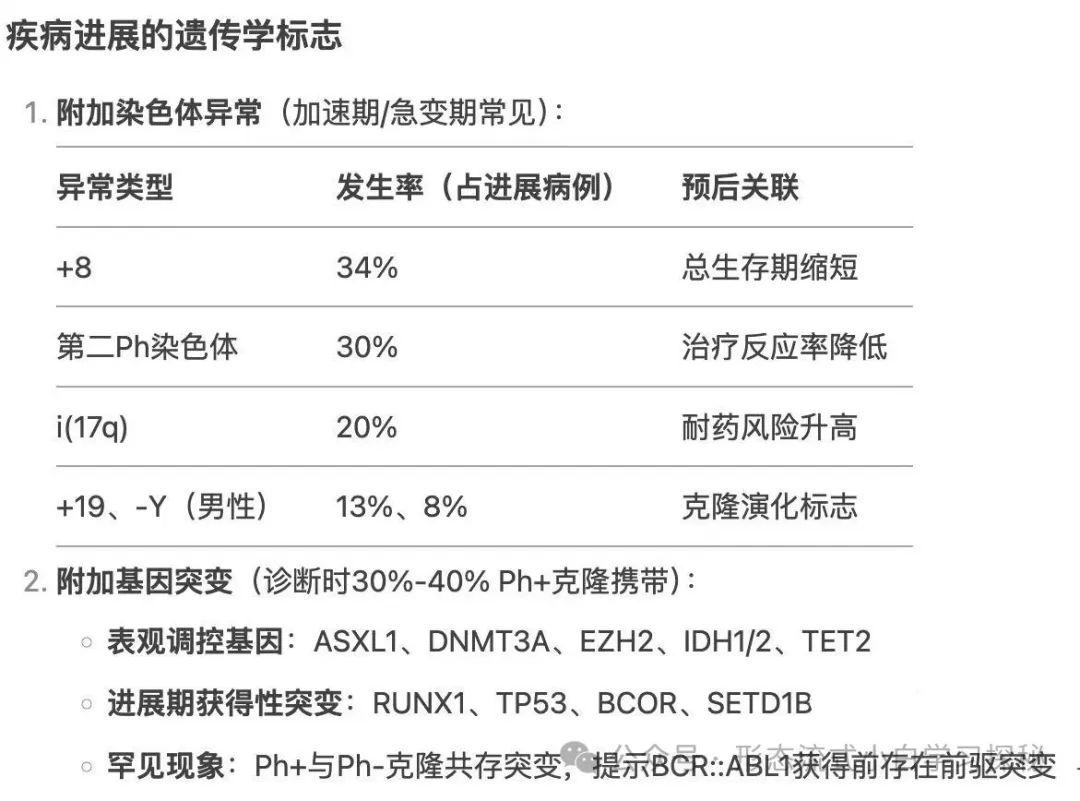
Prognosis and Clinical Commentary
Tyrosine kinase inhibitors (TKIs) have significantly improved the survival and quality of life of patients with Chronic Myeloid Leukemia (CML) by targeting the constitutively active kinase domain of the BCR::ABL1 fusion protein. Today, most newly diagnosed CML patients can expect a life expectancy nearly comparable to that of the normal population after receiving TKI therapy. In the current era of TKI therapy, the most important prognostic indicators are the patient’s response to treatment at the hematological, cytogenetic, and molecular levels. Overall, the complete cytogenetic response rate for first-line TKI therapy can reach 70% to 90%, with 5-year progression-free survival and overall survival rates ranging from 80% to 95%. By 10 years, survival rates can still be maintained at 82% to 85%. Achieving major molecular remission (MMR: BCR::ABL1 <0.1% under international standards) and deeper molecular responses (BCR::ABL1 <0.01% or lower) occurs more rapidly and frequently, with resistance rates to second-generation TKIs being less than 10%. The key to successful TKI therapy lies in the regular and continuous assessment of the patient’s hematological, cytogenetic, and molecular status to timely detect changes indicating drug failure or resistance. In patients receiving TKI therapy, the morphological features of blood and bone marrow samples reflect changes in cytogenetic and molecular events during the treatment response process: white blood cell counts normalize, and the morphology of bone marrow cells tends to normalize, a process typically completed within 3 to 6 months. Although morphological features are insufficiently sensitive to reflect early increases in BCR::ABL1 transcript burden that usually indicate drug resistance, in follow-up samples from patients receiving TKI therapy, if any morphological features suggestive of CML persist or reappear, caution should be exercised regarding drug resistance, and further assessment of cytogenetic or molecular status is warranted. For CML-CP patients with tyrosine kinase domain mutations or BCR::ABL1 amplification, increasing the dose or switching to a new generation of TKIs can prevent or reverse disease progression. Recently, a class of small molecules targeting the ABL1 myristoyl pocket has been introduced into the treatment of CML.