Protein complexes are key molecules that perform complex functions within cells. Understanding the structure of these complexes not only helps to reveal their functional mechanisms but also provides new ideas for drug development and disease treatment. However, with the deepening of research, the number and complexity of protein complexes have increased dramatically, and how to quickly and accurately compare and analyze these structures has become a pressing issue.
Traditional methods for comparing protein complexes face challenges such as high computational load and long processing times, especially when handling large databases. The balance between computational speed and accuracy has always been a difficulty in research. To address this issue, on February 5, Nature Methods published a research report titled “Rapid and Sensitive Protein Complex Alignment with Foldseek-Multimer,” in which researchers developed Foldseek-Multimer, a brand new tool for protein complex comparison. Foldseek-Multimer combines efficient chain-to-chain comparison and superposition clustering techniques, achieving a speed increase of 3 to 4 orders of magnitude over existing traditional tools while maintaining a high level of comparison accuracy.
The core advantage of this breakthrough technology is that it can perform comprehensive comparisons of millions of protein complexes in an extremely short time. When processing large datasets like the PDB database, the efficient algorithm of Foldseek-Multimer not only accelerates the comparison process but also ensures that structural similarities can still be detected even with low sequence similarity. This way, Foldseek-Multimer provides researchers with a new tool that can complete comparisons of billions of complexes in just a few hours, opening up new prospects for protein complex research and drug target discovery.
With the continuous advancement of prediction tools like AlphaFold, predicting the three-dimensional structures of protein complexes has become increasingly accurate. The emergence of Foldseek-Multimer further accelerates the validation and application of these prediction results. Its speed and efficiency allow us to explore the diversity and complexity of protein complexes more deeply, providing important technical support for unlocking the secrets of life.

Protein Complexes: The “Factories” of Life
Within cells, protein complexes are the core “factories” that perform various biological functions. These complexes usually consist of multiple protein subunits that work together through interactions to complete key biological processes such as gene expression, cell signaling, and immune responses. The structure and function of each protein complex highly depend on the precise pairing and spatial arrangement of its subunits. For instance, the polymerase complex involved in DNA replication consists of multiple protein subunits, ensuring that DNA can be replicated efficiently and accurately. Similarly, the immune response of cells often relies on complex antibody-antigen binding mechanisms, where protein complexes play a decisive role.
With the advancement of technology, researchers have been able to resolve the structures of an increasing number of protein complexes using high-resolution techniques such as X-ray crystallography, nuclear magnetic resonance (NMR), and cryo-electron microscopy (cryo-EM). However, despite these techniques providing important structural information, the dramatic increase in the number of protein complexes has posed new challenges in systematically comparing and understanding these structures.
The complexity of protein complexes is not only reflected in their composition of multiple subunits but also in their ability to change structure and function under different physiological conditions. For example, some complexes may undergo dynamic conformational changes during intracellular signaling, directly affecting their biological functions. For instance, the binding of a receptor to a ligand can trigger conformational changes in the receptor, thereby activating downstream signaling pathways. Therefore, understanding these complex structures and functions requires us to not only grasp the structure of individual proteins but also reveal their interactions and synergistic effects within complexes.
The Challenges of Protein Structure Comparison
The structural comparison of protein complexes is a crucial task in biological research, especially when exploring the similarities and differences between different protein complexes. However, traditional comparison methods face significant challenges. One of the main issues is the enormous computational load, particularly when comparing complex multi-chain protein complexes, where traditional methods often require a substantial amount of time and computational resources. For example, when using the US-align tool to compare 931 pairs of protein complexes, the computation on a single-core server took as long as 13 days. Clearly, as the number of protein complexes in databases increases, the speed limitations of traditional methods become increasingly evident.
Moreover, protein complex comparison not only relies on the precise alignment of structures but also needs to solve the problem of correctly pairing each protein chain. Since protein complexes typically consist of multiple subunits, correct chain pairing is key to ensuring comparison accuracy. However, due to the complexity and diversity of protein structures, particularly at low sequence similarity, accurate chain pairing becomes especially challenging. For instance, the US-align method employs a greedy search strategy to propose candidate chain pairings and optimizes them using dynamic programming, which somewhat improves comparison speed but still struggles to break through the bottleneck in large-scale database comparisons.
This contradiction between speed and accuracy renders traditional comparison methods inadequate when facing large datasets of protein complexes. To improve comparison efficiency, many studies have attempted to reduce the number of complexes to be compared through pre-screening steps. For example, the QSalign method reduces the number of complexes to be compared based on sequence similarity, thus accelerating the comparison process. However, while this pre-screening approach can save computational time, it may sacrifice sensitivity, especially in low sequence similarity ranges, potentially missing some structural similarities.
In the challenges of protein complex comparison, the balance between speed and accuracy becomes a core issue. How to improve comparison speed while ensuring the accuracy of the results has always been a focal point for researchers. With the advent of Foldseek-Multimer, this issue has been addressed. Foldseek-Multimer utilizes efficient chain-to-chain comparison and superposition clustering algorithms to significantly enhance comparison speed.
Foldseek-Multimer: A New Revolution in Protein Comparison
First, the core technology of Foldseek-Multimer lies in the combination of efficient chain-to-chain comparison and superposition clustering algorithms. Traditional protein structure comparison methods require processing each pair of protein chains individually, while Foldseek-Multimer dramatically increases computational efficiency through Foldseek’s fast chain-to-chain comparison algorithm. Its uniqueness is that Foldseek-Multimer represents each chain-to-chain comparison as superposition vectors, which record the rotational and translational information of chain alignments. This allows the comparison process to move beyond simple pairwise comparisons and efficiently handle large-scale complex comparisons through superposition clustering algorithms.
Using the DBSCAN (Density-Based Spatial Clustering of Applications with Noise) algorithm, Foldseek-Multimer can iteratively cluster these superposition vectors, identifying structurally similar complexes and quickly calculating optimal comparison results. Notably, the DBSCAN algorithm does not require pre-setting the number of clusters and can automatically adjust clustering parameters based on data distribution, greatly enhancing the flexibility and accuracy of comparisons. In this way, Foldseek-Multimer can complete comparisons of millions of protein complexes in a short time, with comparison quality comparable to traditional methods.
The speed improvement of Foldseek-Multimer is particularly significant. Researchers compared Foldseek-Multimer with the traditional tool US-align, and the results showed that Foldseek-Multimer was over 100 times faster than US-align when performing the same comparison of 931 pairs of protein complexes. When facing large databases, the advantages of Foldseek-Multimer become even more apparent. For example, it can complete the comparison of 5.7 billion pairs of complexes in 11 hours, while traditional methods might take months to complete. This breakthrough makes the work of comparing protein complexes more efficient and greatly accelerates the progress of structural biology research.
Additionally, Foldseek-Multimer demonstrates extremely high sensitivity when handling complexes with low sequence similarity. During the comparison process, Foldseek-Multimer not only relies on sequence similarity but also identifies potential structural similarities through matching structural information. For instance, when using Foldseek-Multimer to compare a CRISPR-Cas system in the PDB database, even though the query complex had a sequence similarity of only 11.1% to 19.8% with similar complexes in the database, Foldseek-Multimer was still able to identify the structural similarities between these complexes and provide effective comparison results. This is especially important because many significant biological discoveries often exist among complexes with low sequence similarity, and the high sensitivity of Foldseek-Multimer ensures that these potential structural similarities are not overlooked.
Through rapid and accurate large-scale comparisons, Foldseek-Multimer not only accelerates the comparison process of protein complexes but also provides researchers with more clues about structural similarities, facilitating the resolution of complex biological issues.
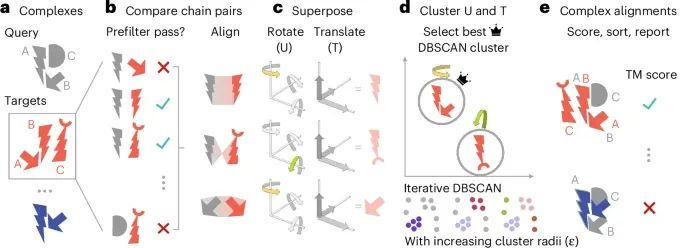
The Working Principle and Process of Foldseek-Multimer in Protein Complex Structure Comparison (Credit: Nature Methods)
Fast Query Function: Foldseek-Multimer can quickly query the input protein complex (or multiple complexes) against a large database that may contain millions of target complexes. Through this method, Foldseek-Multimer can effectively narrow down the comparison range, enhancing comparison efficiency.
Chain-to-Chain Comparison: Each chain in the image (gray) will be compared with each chain in the target complex (red). To improve computational efficiency, Foldseek-Multimer uses a pre-screening function that can quickly exclude non-matching chain pairs, ensuring that only potential structurally similar complex pairs undergo comprehensive comparison.
Superposition Alignment Representation: Foldseek-Multimer represents each chain-to-chain comparison as superposition, achieving docking of the target chain with the query chain through rotation and translation. In a simplified example, two chain-to-chain comparisons (upper and lower) rotate along the same axis (highlighted in yellow and green), while the middle comparison rotates along different axes. This process illustrates how to achieve precise alignment of chains through geometric transformations.
Complex-to-Complex Comparison: By integrating multiple chain-to-chain comparisons, Foldseek-Multimer derives the final comparison results between complexes. In this process, the similarity of chain-to-chain superposition representations is used to determine the quality of complex comparisons. Foldseek-Multimer utilizes the DBSCAN algorithm for iterative clustering, identifying the best superposition clustering by gradually increasing the clustering radius to calculate the optimal comparison between complexes.
TM Score Calculation: After selecting the best scoring cluster, Foldseek-Multimer calculates the TM score between complexes based on that cluster. The TM score assesses the structural similarity between complexes by evaluating the results of all chain-to-chain comparisons between the query complex and the target complex.
Integration with AlphaFold: A New Era in Protein Structure Prediction
With the emergence of protein structure prediction tools such as AlphaFold, predicting the three-dimensional structures of proteins has entered a new era. AlphaFold can predict the three-dimensional structures of single-chain proteins with remarkable accuracy using deep learning technology, while Foldseek-Multimer provides robust support, especially in the structural comparison and validation of protein complexes. The combination of the two not only accelerates the progress of protein structure research but also offers new perspectives for exploring protein functions and interactions.
The advent of AlphaFold provides a powerful tool for protein structure prediction, demonstrating unprecedented accuracy in predicting the structures of single-chain proteins. However, predicting complex protein complexes remains challenging. Complexes typically involve multiple subunits, and the interactions between them, as well as conformational changes in different environments, exponentially increase the complexity of predictions. Therefore, while AlphaFold can predict the structure of a single protein, precise structure predictions for multiple protein subunits require further validation and comparison tools to confirm the rationality of these structures.
At this point, the role of Foldseek-Multimer becomes particularly prominent. Through its efficient protein complex comparison function, Foldseek-Multimer can quickly validate the structures of protein complexes predicted by AlphaFold. For example, in the case of the CRISPR-Cas complex predicted by AlphaFold, researchers used these prediction results as queries and employed Foldseek-Multimer to compare them with known structures in the database. In this experiment, Foldseek-Multimer was not only able to quickly identify structurally similar complexes but also confirm the correctness of the predicted structures. This combination allows the prediction of protein complex structures to no longer rely solely on computational predictions but rather to be validated through real structural data, thereby improving the accuracy and reliability of predictions.
Furthermore, the integration of Foldseek-Multimer with AlphaFold will drive the forefront development of protein complex research. With the high efficiency of Foldseek-Multimer’s comparison capabilities, researchers can quickly compare and validate the predicted structures of millions of protein complexes. For instance, in comparisons with the PDB database, Foldseek-Multimer can complete comparisons of 5.7 billion pairs of complexes in just a few hours, helping researchers filter out the most relevant complex structures. This efficient and accurate comparison not only saves a significant amount of computational time but also makes large-scale protein complex research feasible.
By combining the structure prediction capabilities of AlphaFold with the efficient comparison algorithms of Foldseek-Multimer, researchers can explore the structures and functions of protein complexes more quickly and accurately. This synergistic effect not only accelerates the discovery of new protein functions but also provides new methods for drug target screening.
Foldseek-Multimer: Accelerating Drug Development and Disease Treatment
As the field of biomedicine continues to develop, drug development faces increasing challenges, particularly in the discovery of new drug targets and the study of disease-related proteins. Traditional drug discovery methods often rely on experimental validation of targets, a process that is both time-consuming and costly. The emergence of Foldseek-Multimer brings unprecedented efficiency to drug development, as it can swiftly identify structural similarities of protein complexes through structural comparison techniques, providing new directions for drug target discovery.
In the drug development process, the selection of targets is key to success. Effective targets are often proteins or protein complexes associated with specific diseases. Foldseek-Multimer, with its efficient comparison algorithms, can assist researchers in finding other complexes similar to target complexes within massive protein databases. For example, when comparing existing protein complexes in the PDB database, Foldseek-Multimer can quickly identify functionally similar protein complexes, providing strong support for potential drug target screening.
Moreover, Foldseek-Multimer can reveal structural similarities between proteins even under low sequence similarity conditions. This is particularly important for studying protein complexes that have low sequence similarity but similar functions. Many disease-related proteins, such as those involved in cancer and neurodegenerative diseases, often exhibit low sequence similarity, making it difficult for traditional methods to discover these potential targets. However, Foldseek-Multimer, with its superposition clustering algorithm, can break through this limitation and help researchers identify potential drug targets across different species and disease states.
Additionally, the potential of Foldseek-Multimer in accelerating the study of disease-related protein complexes is immense. When researching complex diseases, researchers typically need to analyze a large number of protein complexes and their interactions to uncover potential mechanisms of the disease. However, traditional comparison methods are often limited by excessive computational loads and slow comparison speeds. The emergence of Foldseek-Multimer greatly accelerates this process. With its efficient comparison capabilities, researchers can perform structural comparisons of thousands of protein complexes in a short time, thereby more swiftly identifying protein complexes potentially related to diseases.
For instance, in the case of viral infections or cancer-related protein complexes, Foldseek-Multimer can quickly compare protein complexes from different patient samples to identify common features. This not only accelerates disease research but also helps scientists pinpoint potential therapeutic targets more accurately, laying a solid foundation for the development of new drugs.
By applying Foldseek-Multimer to drug development and disease research, researchers can more efficiently screen potential targets, shorten drug development cycles, and increase the success rate of drug development. This technology undoubtedly brings revolutionary changes to the field of biomedicine, providing unprecedented opportunities to tackle various diseases, especially complex ones. As Foldseek-Multimer continues to optimize, its application potential in drug discovery and disease treatment will be further unleashed, becoming an important tool in the field of biomedicine.
The future potential of Foldseek-Multimer is not limited to existing protein complex research. As the structural study of RNA and DNA complexes progresses, the technical framework of Foldseek-Multimer can be extended to these new research areas. For example, RNA and DNA also exist in various complex forms, and their structures and functions are equally complex and critical. Foldseek-Multimer can draw on its successful experience in protein complexes to provide new solutions for the comparison and structural analysis of RNA and DNA complexes. This expansion will further promote research in molecular biology, genomics, and cell biology. Foldseek-Multimer not only changes the way protein complexes are compared but also offers new perspectives and technical means for future biological research.
Kim W, Mirdita M, Levy Karin E, Gilchrist CLM, Schweke H, Söding J, Levy ED, Steinegger M. Rapid and sensitive protein complex alignment with Foldseek-Multimer. Nat Methods. 2025 Feb 5. doi: 10.1038/s41592-025-02593-7. Epub ahead of print. PMID: 39910251.
Please cite the source from 【Biological Exploration】
Statement: This article is for sharing purposes only and does not represent the platform’s position. If there are copyright issues, please contact us as soon as possible, and we will correct it immediately. Thank you!
Science | Why Are T Cells “Exhausted”? Answers from Metabolism and Epigenetics
Nature Biotechnology | New Breakthroughs in Immunotherapy: How to Precisely Target Diseased Cells?
Cell | Breaking Traditional Cognition: How Electrical Synapses Dynamically Regulate Neural Network Behavior
Nature Biotechnology | How to Decode the “Genomic Dark Matter” of Non-coding Regions?
Nature Methods | How to Precisely Manipulate Cells Using Temperature? New Ideas for Cell Fate Regulation