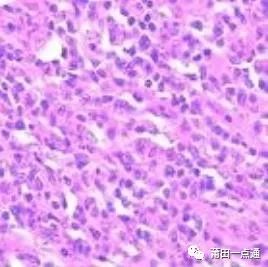
Diffuse Large B-Cell Lymphoma: Composed of centroblastic (non-cleaved), B immunoblastic, and anaplastic large B-cell lymphomas classified by Kiel. Under the microscope: a relatively uniform morphology of large cells arranged diffusely can be seen. The cell morphology is diverse, resembling centroblasts, immunoblasts, and may also include multinucleated tumor cells with anaplastic features.
Incidence Rate
Diffuse large B-cell lymphoma is currently the most common adult non-Hodgkin malignant lymphoma, accounting for 30-40% of adult non-Hodgkin malignant lymphomas in Western countries, with an even higher proportion of 60% in developing countries. In childhood lymphomas, DLBCL accounts for less than 10%. The age range for DLBCL onset is wide, with an average age of 70, but it can also occur in children; male patients outnumber female patients.
WHO Definition
The definition of diffuse large B-cell lymphoma is that large tumor B lymphocytes grow diffusely, with the nuclei of tumor cells being similar to or larger than those of normal tissue cells, and cell sizes being at least twice that of normal lymphocytes.
Related Names
Rappaport Classification: Diffuse histiocytic type, diffuse lymphocytic and histiocytic mixed type
Kiel Classification: Centroblastic type, B immunoblastic type, anaplastic large B-cell type
Lukes-Collins Classification: Large cleaved follicular center cell type, large non-cleaved follicular center cell type, B immunoblastic type
Working Formulation Classification: Diffuse large cell type, large cell immunoblastic type, diffuse mixed small and large cell type
REAL Classification: Diffuse large B-cell lymphoma
Etiology and Pathogenesis
The etiology of diffuse large B-cell lymphoma is currently unclear. It is usually primary but can also progress or transform from low-grade lymphomas (such as follicular lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, marginal zone B-cell lymphoma, and nodular lymphocyte predominant Hodgkin lymphoma). Some cases occur against a background of autoimmune diseases or immunodeficiency.
Pathological Histology
Typical DLBCL shows diffuse proliferation of tumor cells that invade the normal structure of affected lymph nodes or extranodal sites. Lymph node involvement can be complete, partial, follicular, sinusoidal, or a mixture of several forms. Extranodal soft tissue and vascular infiltration are common, with widespread or clear sclerosis observed (some cases are accompanied by significant sclerosis, forming partitioned nodules or “Indian file” phenomenon). Tumor cells are large transformed lymphocytes, with sizes varying greatly between different cases or within the same case, with nuclei larger than those of reactive tissue cells. However, in some cases, nuclei may be medium-sized, making differentiation from Burkitt-like lymphoma difficult. Nuclei are round, serrated, or irregularly folded, with vacuolated or coarse granular chromatin, often with nucleoli of varying sizes, eosinophilic or basophilic, one or more. Cytoplasm is moderate or abundant, can be transparent, lightly stained, or eosinophilic. In some cases, tumor cells show plasmacytoid features: eosinophilic, with pale-stained perinuclear Golgi halos. Eosinophilic cytoplasmic fragments may be present, indistinguishable from “plasma cell bodies” in inflammatory responses. Multilobular cells or atypical cells resembling Reed-Sternberg cells may be observed. Mitoses are frequently seen. From a cytological perspective, the morphology of tumor cells is diverse and can be further classified morphologically, but there are no significant differences in immunophenotype or genetic characteristics between subtypes. Therefore, a unified diagnosis of diffuse large B-cell lymphoma can be used alongside morphological classification.
Subtypes
In the WHO classification, diffuse large B-cell lymphoma is divided into four variants based on histological morphological changes: centroblastic type, immunoblastic type, T-cell/histiocyte-rich type, and anaplastic type, with two additional rare subtypes: mediastinal sclerosing large B-cell lymphoma and intravascular lymphoma.
Centroblastic Type
(centroblastic type)
This can be a centroblastic lymphoma composed of a single morphology of multinucleated cleaved cells, or a characteristic polymorphic cell infiltration formed by a mixture of centroblast-like cells and multinucleated cleaved cells.
Tumor cells range from medium to large lymphocytes, oval or round. The nuclei are vesicular, with fine chromatin, and 2-4 nucleoli located beneath the nuclear membrane. Cytoplasm is sparse, eosinophilic or basophilic.
Immunoblastic Type
(Immunoblastic)
More than 90% of tumor cells are immunoblasts. A centrally located nucleus with scant eosinophilic cytoplasm. Some cells show plasmacytic differentiation. Its clinical and immunophenotypic characteristics help differentiate it from the plasmacytic differentiation type of plasmacytoma.
T-Cell / Histiocyte Rich Type
(T-cell / histiocyte rich)
Large tumor B cells are less than 10%, with most cells being reactive T cells, and histiocytes may be present or absent. This type is characterized by isolated or clustered large lymphoma cells scattered among many small lymphocytes (small, round or slightly larger, irregularly elongated nuclei). Histiocytes, epithelial-like cells, eosinophils, and plasma cells may also be mixed in. Small venules rich in endothelial cells are prominent.
Large tumor cells can be L&H cells, centroblasts, immunoblasts, or Reed-Sternberg cells. B-cell markers are expressed, and genetic analysis further confirms the monoclonal proliferation of B cells. Recent studies suggest that tumor cells may originate from germinal center B cells.
Anaplastic Type
(anaplastic)
Tumor cells are large, round, oval, or polygonal, with pleomorphic nuclei resembling Reed-Sternberg cells. They may grow in a cancer nest-like manner or sinusoidal growth. Tumor cells express EMA positively, while LCA may be negative. This type of tumor differs biologically and clinically from anaplastic large T-cell lymphoma.
Genetic Classification
With the development of immunology research and molecular biology techniques, especially the advent of DNA microarray technology, gene expression profiling analysis of a large sample of diffuse large B-cell lymphoma cases has classified tumor cells into two categories based on different gene expression patterns: germinal center B-cells and in vitro activated peripheral blood B-cells. This divides diffuse large B-cell lymphoma into germinal center-like B-cell and activated B-cell-type B-cell lymphomas. Recent research by Andreas Rosenwald et al. has discovered a third type of tumor cell that does not highly express the characteristic genes of the above two cell types, naming this type of tumor Type 3 diffuse large B-cell lymphoma (Type 3 DLBCL). Numerous studies have shown that the germinal center B-cell type has the best prognosis, while the activated B-cell type has the worst.
Cellular Immunohistochemistry
Diffuse large B-cell lymphoma expresses various pan-B-cell markers such as CD19, CD20, CD22, CD79a, and BSAP (B-cell specific activator protein, encoded by the PAX5 gene), but may lose one or more of them. HLA-DR is often positive. Some cases express activated lymphocyte markers such as CD25 and CD30. 50-75% of cases can detect surface and/or cytoplasmic immunoglobulin (IgM > IgG > IgA), with cytoplasmic Ig being common in cases with plasmacytic differentiation. The vast majority of anaplastic large B-cell lymphomas have CD30 protein expression, but non-anaplastic types may also occasionally show CD30 staining. 10% of cases express CD5, and 25-50% express CD10. Studies indicate that CD5-positive DLBCL is more likely to be considered a primary tumor rather than a progression from SLL/CLL, and this type of lymphoma lacks Cyclin D1 expression, helping to differentiate it from the plasmacytic variant of mantle cell lymphoma. About 30-50% of cases express Bcl-2 protein; a high proportion of cases show nuclear expression of Bcl-6 protein. A minority of cases may express P53 protein, usually associated with P53 gene mutations. A few cases may express plasmacytic-related markers, such as Syndecan (CD138). The expression positivity rate of Ki-67 is generally over 40%, with a few cases exceeding 90%.
Molecular Biology
From a genetic research perspective, DLBCL is a tumor with multiple genetic actions, showing genetic heterogeneity. Many cases exhibit complex genetic abnormalities.
Most cases show clonal rearrangements of immunoglobulin heavy and light chain genes, with mutations in the variable regions. 20-30% of cases may exhibit translocations of the Bcl-2 gene, a marker of follicular lymphoma. Over 30% of cases show abnormalities in the 3q27 region, affecting the primary oncogene Bcl-6. Myc gene rearrangement is uncommon. Some cases may detect EB virus infection, especially in those with underlying immunodeficiency. Recent studies using cDNA microarray technology to detect gene expression patterns have defined two major molecular categories, suggesting different stages of B-cell development. One type has the expression profile characteristic of germinal center B-cells, while the other resembles the expression profile of in vitro activated peripheral blood B-cells, indicating different tumor cell origins.
Diagnosis and Differential Diagnosis
In routine external examination work, diffuse large B-cell lymphoma needs to be differentiated from the following diseases:
Metastatic Carcinoma or Malignant Melanoma
Differentiating large cell lymphoma from metastatic carcinoma or malignant melanoma morphologically poses certain difficulties; immunomarkers are the best solution to this issue, but appropriately selecting antibodies for marking is very important. For example, in ALCL-type DLBCL, 30% of cases may not express LCA, and 60% express EMA. Without sufficient comparison, antibody marking may mislead the diagnosis.
Infectious Mononucleosis
Infectious mononucleosis shows very active immunoblastic proliferation, making it difficult to differentiate from large cell lymphoma. Similar vigorous reactive lymphoid proliferation can also be seen in other viral infections and allergic reactions. The following clues can help diagnose infectious mononucleosis in cases suspected to be large cell lymphoma:
(1) Patient’s age: diagnosing large cell lymphoma in children or adolescents should be done with great caution, leaning more towards infectious mononucleosis;
(2) Lymph node structure is not completely lost, with parts showing obvious sinuses and reactive follicles (necrosis may occur), leaning towards infectious mononucleosis;
(3) Immunoblasts lack certain atypical features (such as obvious nuclear irregularity), indicating the need to consider infectious mononucleosis.
Necrotizing Lymphadenitis
Necrotizing lymphadenitis is a self-limiting lymphadenitis, usually occurring in young patients, commonly in the neck, with active proliferation of activated lymphocytes, showing obvious nuclear irregularity, easily misdiagnosed as large cell lymphoma. If activated lymphocytes appear around the necrotic focus, lacking involvement of surrounding lymph node tissues, with abundant nuclear debris and many macrophages, crescent-shaped nuclei support the diagnosis of necrotizing lymphadenitis rather than large cell lymphoma.
Burkitt Lymphoma and Extramedullary Leukemia
Typical Burkitt lymphoma is easily differentiated from DLBCL, but in Eastern DLBCL, Burkitt-like differentiation may occur, with tumor cells being medium-sized or smaller, uniform, with phagocytic histiocytes being common, easily misdiagnosed as Burkitt lymphoma. However, careful examination will still reveal some slightly larger cells with germinal center blast differentiation and more significant phagocytic histiocyte proliferation forming starry sky phenomena, with high EBV expression also being an important auxiliary indicator. At the same time, the following features can be used to diagnose non-Burkitt lymphoma.
(1) Mitoses and nuclear debris are common;
(2) Cytoplasm and nucleus are square or cast-like;
(3) Cell proliferation index (Ki-67) ≥80%;
(4) CD10 positive;
(5) Leu-8 negative.
Acute myeloid leukemia may present as a solid tumor in some cases, such as occurring in the orbit, intestinal wall, or uterus, termed granulocytic sarcoma. Histologically, the cells are large, with scant cytoplasm, large nuclei, and round morphology, easily diagnosed as lymphoma, but immunomarkers do not support this. Aside from LCA positivity, no T/B cell markers are expressed. Careful differentiation from extramedullary leukemia is required, with further markers such as lysozyme (Lys), myeloperoxidase (MPO), and α1-antitrypsin also being expressed. Morphologically, attention should be paid to the presence of eosinophilic and neutrophilic granules in the cytoplasm, and the kidney or bean-shaped morphology of the nuclei should raise high suspicion for granulocytic sarcoma. DLBCL is rare in the orbit and nervous tissue.
Anaplastic Large Cell Lymphoma
Morphologically difficult to distinguish, requiring immunohistochemical staining. Strictly speaking, anaplastic large cell lymphoma is a T-cell or null-cell lymphoma, so it should express T-cell surface markers or non-T non-B expressions. If B-cell markers are expressed, it may be a variant of DLBCL.
Hodgkin Lymphoma (HD)
B-cell lymphoma types rich in T-cells in DLBCL morphologically resemble the nodular lymphocyte predominant type of HD, but large cells usually do not have the large nucleoli of H or Reed-Sternberg cells, with extremely irregular nuclei. Large cells respond to LCA and B-cell markers, supporting the diagnosis of B-cell lymphoma, especially when monoclonal Ig is present, confirming this diagnosis. Caution is needed when determining CD20 staining results, as Reed-Sternberg cells can also be positive in heterogeneous HD types.
Nodular Sclerosis Hodgkin Lymphoma
Nodular sclerosing Hodgkin lymphoma (NSHD) is the most common subtype of HD in the West. This type poses challenges in differential diagnosis. Its characteristics include nodular growth patterns and the formation of collagen bands within nodules with the appearance of lacunar cells. There is significant eosinophilic infiltration, even forming eosinophilic abscesses. The NSHD mixed cell subtype is characterized by lacunar cells and mononuclear Reed-Sternberg cells adhering to form sheets, with relatively few reactive lymphocytes, resembling DLBCL, but the presence of numerous multinucleated Reed-Sternberg cells and lacunar cells should alert to HD. The mixed cellularity (MC) subtype of HD is predominant in China, and abundant histiocytic proliferation should not be misidentified as tumor cells. Meanwhile, immunohistochemistry helps differentiate, as HD tumor cells are usually LCA(-), CD15(+), and 40-60% of cases test EBV positive; DLBCL is usually LCA(+), CD15(-), and the combined application of B and T cell immunomarkers has diagnostic value.
Mediastinal (Thymic) Large B-Cell Lymphoma
(Mediastinal/thymic large B-cell lymphoma)
This is a subtype of diffuse large B-cell lymphoma that occurs in the mediastinum and is currently thought to originate from thymic B cells, with unique clinical, immunophenotypic, and genetic features. It is highly prevalent in individuals aged 30-40, more common in females. Clinically, it often presents as a localized mass in the anterior mediastinum, sometimes accompanied by superior vena cava syndrome, and can involve extranodal sites such as the kidneys, adrenal glands, liver, skin, and brain. The etiology is unknown, and EBV infection has not been detected.
Morphologically, it presents as a variable number of fibrous septa infiltrating tumor cells diffusely. Sometimes, residual thymic tissue appears in small clusters, easily confused with sarcoma. Differences in tumor cell size and nuclear shape between different cases and different regions of the same case are significant. Most tumor cells have abundant, lightly stained cytoplasm. Some cases may be accompanied by reactive lymphocytes and eosinophilic infiltration, easily misdiagnosed as Hodgkin lymphoma. Rarely, it may be associated with nodular sclerosing Hodgkin lymphoma (composite lymphoma). However, due to the tumor occurring in the mediastinum, biopsy specimens are scarce, and due to the common occurrence of collagen sclerosis, many artificial artifacts can complicate diagnosis.
Tumor cells express B-cell markers such as CD19 and CD20, while Ig, HLA class I and II molecules are rarely detected, with no expression of CD10 or CD5. Tumor cells usually express CD30 protein, but weakly; CD30 expression can be widespread or limited to local areas. Tumor cells express LCA (CD45), which classic Hodgkin lymphoma does not express. Immunoglobulin gene rearrangements can be detected, even when Ig protein expression is absent. Hyperdiploid karyotypes (located at the 9th chromosome arm) and REL gene amplification are often detected, indicating this subtype is distinctly different from diffuse large B-cell lymphoma occurring in other sites. Recently, a high proportion of cases have been found to have overexpression of the MAL gene. Tumor cells do not show rearrangements of BCL2, BCL6, or MYC genes.
Intravascular Large B-Cell Lymphoma
(Intravascular large B-cell lymphoma, angiotropic lymphoma)
Previously referred to as “tumor-associated vascular endothelial disease,” it is a special subtype of extranodal diffuse large B-cell lymphoma. The tumor cells are transformed peripheral blood B cells. Its characteristic is that tumor cells infiltrate only within the lumen of small blood vessels, with some growing within capillaries. It is speculated that this unique growth pattern may be related to defects in tumor cell homing receptors; recent studies have shown that in intravascular large B-cell lymphoma, the expression of CD29 (β1 integrin) and CD54 (ICAM-1) adhesion molecules is lacking. Most cases may present with abnormal neurological symptoms or skin lesions but can also invade systemic organs. The disease progresses rapidly, often leading to death, but some cases have been reported to achieve complete remission after appropriate chemotherapy.
Grossly, the tumor often presents as extensive tissue hemorrhage, thrombosis, and necrosis, with no visible tumor mass. Histological features include large tumor cells, loosely arranged, with vesicular nuclei, clear nucleoli, and frequent mitotic figures; in very few cases, anaplastic tumor cells may be seen. Tumor cells invade the lumen of small blood vessels, and some cases may develop fibrinous emboli. Vascular occlusion often leads to widespread infarction, and sometimes lymphoma cell components can be seen outside the vessels. When the tumor invades the lungs and bone marrow, the infiltration may be very subtle. Meanwhile, CD45 and CD20 immunohistochemical staining can be used to distinguish single tumor cells within capillaries. Tumor cells are rarely found in cerebrospinal fluid and blood.
Tumor cells usually express B-cell-associated antigens (such as CD19, CD20, CD22, CD79a). Individual cases may co-express CD5. Intravascular T-cell lymphoma is extremely rare. Endothelial cell-associated antigen factor VIII can be detected, but it is generally considered not to be expressed by tumor cells but rather an adsorption effect of factor VIII. Most cases show immunoglobulin gene rearrangements, while T-cell receptor gene rearrangements are rare. Abnormal karyotypes can be detected, but due to the small number of cases, it is difficult to show a more uniform pattern.
Primary Effusion Lymphoma
(Primary effusion lymphoma, PEL)
When large B-cell lymphoma manifests as serous effusion rather than a solid tumor, it is termed primary effusion lymphoma (PEL), originating from post-germinal center B cells. It is associated with human herpesvirus 8 (HHV-8) and Kaposi’s sarcoma-associated herpesvirus (KSHV) infection, mostly seen in HIV-infected patients, predominantly middle-aged homosexual males. However, this lymphoma is very rare even among HIV-infected individuals. It has also been found in non-HIV-infected patients who have undergone allogeneic transplantation. Some patients without immunodeficiency, especially elderly males in regions with high HHV-8/KSHV infection (such as the Mediterranean region), have also been reported. This type of lymphoma has a poor prognosis, with a median survival of less than 6 months.
The most commonly affected sites are the pleura, pericardial cavity, and peritoneal cavity. A typical patient may have only one body cavity involved. The gastrointestinal tract, soft tissues, and other extranodal organs may also be affected. The typical clinical symptom is the absence of lymphadenopathy and organic tumor (organomegaly), presenting only as effusion. Some cases have a history of Kaposi’s sarcoma, and very few cases may be related to multicentric Castleman disease.
All cases are HHV-8/KSHV positive. Most cases are associated with EB virus infection. In effusion lesions, high levels of CK can be monitored, especially IL-6 and IL-10.
After centrifugation of the effusion, Wright or May Grunwald Giemsa staining is performed for observation, with tumor cells varying in size, showing immunoblastic or plasmablastic differentiation, and may also have anaplastic differentiation. Tumor cells have large, round or irregular nuclei, with prominent nucleoli. Cytoplasm is abundant, and individual cells may have eosinophilic vacuoles. In plasmacytic differentiated tumor cells, perinuclear halos can be seen. Some cells resemble Reed-Sternberg cells. In contrast, histological sections show more consistency in tumor cell morphology. However, tumor cells are generally larger and morphologically diverse. Pleural biopsies show tumor cells adhering to the pleural surface, mixed among fibrinous material, occasionally infiltrating the pleura. This needs to be differentiated from empyema associated with diffuse large B-cell lymphoma, which often has pleural masses damaging the pleura. Tumor cells are EBV positive, HHV-8/KSHV negative.
Tumor cells usually express LCA but generally do not express pan-B-cell markers such as CD19, CD20, and CD79a; surface and cytoplasmic Ig are also absent. There may usually be expression of activated lymphocyte and plasma cell markers such as CD30, CD38, and CD138. Abnormal cytoplasmic expression of CD3 protein may occur, making immunotyping sometimes difficult to determine. Tumor cells may have nuclear expression of HHV-8/KSHV-related latent proteins, which is of significant diagnostic importance. Although EB virus infection is common, LMP-1 staining is usually negative. There are immunoglobulin gene rearrangements and mutations, and some cases also have T-cell receptor gene rearrangements, with no characteristic chromosomal abnormalities detected. Comparative genomic analysis shows that this type of tumor shares similar sequences on chromosome 12 and the X chromosome with other HIV-associated lymphomas. All cases can detect HHV-8/KSHV viral genomic sequences. Most cases are EBER in situ hybridization positive, especially in those elderly patients who are HIV negative.
Disclaimer: We respect originality, and the main purpose is to share information. This article’s text and images are all reprinted from the internet, and copyright belongs to the original author. If any infringement of your rights occurs, please inform us in a timely manner, and we will delete it within 24 hours. We maintain neutrality regarding the content and views expressed in the article and do not provide any express or implied guarantees of its accuracy, reliability, or completeness, for reference only.