
Click the blue text
Follow us

Primary myelofibrosis (PMF) has made significant progress in diagnosis and treatment in recent years, such as updates to the WHO diagnostic criteria, new clinical trial data for ruxolitinib, and its market approval in China. To provide standardized clinical practice guidance for hematology doctors in China regarding PMF, the Leukemia and Lymphoma Group of the Hematology Branch of the Chinese Medical Association has led the formulation of this guideline based on the “Expert Consensus on the Diagnosis and Treatment of Primary Myelofibrosis in China (2015 Edition)”.
I. Diagnosis Procedures
(1) Medical History Collection
It is essential to carefully inquire about the patient’s age, history of thromboembolism, cardiovascular risk factors (such as hypertension, hyperlipidemia, diabetes, smoking, and congestive heart failure), symptoms such as fatigue, early satiety, abdominal discomfort, skin itching, and bone pain, as well as their activity level, attention, weight loss in the past year, unexplained fever or severe night sweats, history of blood product transfusions, and any family history of similar diseases. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF-TSS, abbreviated as MPN-10)[2] should be used to assess the symptom burden of the patient.
(2) Laboratory Tests
The following laboratory tests are essential for suspected PMF patients[3-8]: ① Complete blood count; ② Bone marrow aspiration smear and peripheral blood smear differential count; ③ Bone marrow biopsy pathological cytological analysis; ④ Chromosome karyotype analysis (±FISH) (if bone marrow is “dry-tapped”, peripheral blood specimens can be used); ⑤ JAK2, MPL, and CALR gene mutation and BCR-ABL1 fusion gene detection (if bone marrow is “dry-tapped”, peripheral blood specimens can be used), ASXL1, TET2, DNMT3a, SRSF2, U2AF1, EZH2, IDH1/2, SF3B1, TP53, and CBL gene mutations as second-line tests; ⑥ Serum erythropoietin (EPO) levels, uric acid, lactate dehydrogenase, liver function, serum iron, ferritin, and other biochemical tests; ⑦ Liver and spleen ultrasound or CT examination, and MRI is recommended for determining spleen volume in qualified institutions; ⑧ HLA typing for patients who may undergo hematopoietic stem cell transplantation (HSCT).
(3) Bone Marrow Biopsy Pathological Cytological Analysis
1. Pathological cytological analysis: The diagnosis of PMF relies on bone marrow biopsy. To ensure accurate pathological analysis, the length of the biopsy tissue should be at least 1.5 cm, and paraffin embedding should be used, with a section thickness of 3-4 μm. Bone marrow biopsy sections should include routine HE and/or Giemsa staining, reticulin (silver staining), PAS staining, chloroacetate AS-D naphthol esterase staining (CE), and Prussian blue staining (iron staining), as well as immunohistochemical staining using CD34 and CD61 monoclonal antibodies.
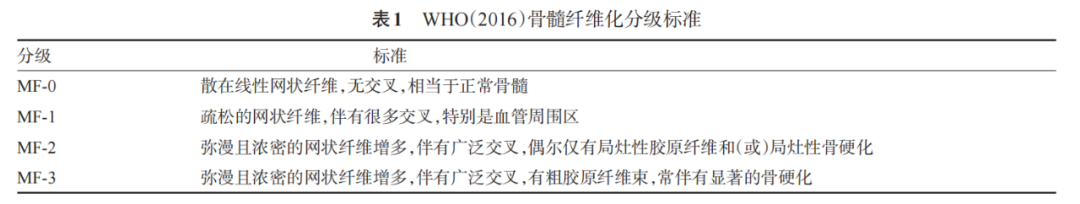
Microscopic analysis should include at least the following: ① Cell proliferation degree: reduced, normal, or active, and whether the proliferation degree corresponds to the patient’s age (20-30 years: hematopoietic tissue 70%-60%; 40-60 years: hematopoietic tissue 50%-40%; 70-80 years: hematopoietic tissue 40%-30%; >80 years: hematopoietic tissue 20%-10%); ② Granulocyte: increased, normal, or decreased, with or without left shift; ③ Erythroid cells: increased, normal, or decreased, with or without left shift; ④ Granulocyte/erythroid ratio (combined with PAS and CE staining); ⑤ Megakaryocytes: increased or decreased, distribution pattern (random distribution, loosely clustered distribution, densely clustered distribution, abnormal distribution near bone trabeculae), cell size, and nuclear morphology (normal/multilobated/reduced lobation/naked nucleus); ⑥ CD34+ cells: proportion (0-9%/10%-19%/≥20%), with or without clustering (>3 cells); ⑦ Fibrosis grade [according to WHO (2016) standards]: fibrosis grade, collagen grade, and bone sclerosis grade; ⑧ Presence of hematopoietic cells in the sinuses. After comprehensive analysis of the above parameters, a possible diagnosis should be made.
2. Bone marrow fibrosis grading standard: Use the WHO (2016) standard[9-10] (Table 1).
II. Diagnosis and Differential Diagnosis
(1) Diagnostic Criteria
Use the WHO (2016) diagnostic criteria[10], which include pre-fibrotic/early PMF (Table 2) and overt fibrotic PMF (Table 3).


(2) Differential Diagnosis
Common causes of reactive bone marrow fibrosis include infections, autoimmune diseases, chronic inflammatory diseases, hairy cell leukemia or other lymphatic tumors, myelodysplastic syndromes (MDS), metastatic tumors, and toxic (chronic) myelopathies.
Pre-fibrotic/early PMF should be differentiated from essential thrombocythemia (ET), primarily relying on pathological histological analysis of bone marrow biopsy[11-12]. In “true” ET patients, the age-adjusted degree of bone marrow proliferation is either absent or slightly elevated, with no significant proliferation of myeloid and erythroid hematopoiesis, and the megakaryocytes show synchronized enlargement of cytoplasm and nucleus, with size ranging from large to giant, and the nucleus is highly lobulated (horn-like), with silver staining fibrosis grade often being MF-0; in pre-fibrotic/early PMF patients, the age-adjusted degree of bone marrow proliferation is significantly increased, with significant proliferation of myeloid hematopoiesis, decreased erythroid hematopoiesis, and the nucleus volume of megakaryocytes increases more than the cytoplasm, with sizes ranging from small to giant, showing clustered distribution, and the nucleus is low lobulated and cloud-like, with silver staining fibrosis grade often being MF-0 or MF-1.
PMF with cytopenia should be differentiated from MDS with bone marrow fibrosis: nearly 50% of MDS patients show mild to moderate reticulin fibrosis (MF-0 or MF-1) in the bone marrow, and 10%-15% of patients show significant fibrosis (MF-2 or MF-3)[13]. Unlike PMF, MDS with bone marrow fibrosis often presents with pancytopenia, and atypical and fragmented red blood cells are less common, with significant three-line developmental abnormalities observed in the bone marrow, and collagen fiber formation is very rare, often without hepatosplenomegaly.
III. Prognostic Assessment Criteria
After confirming PMF, patients should be grouped according to the International Prognostic Scoring System (IPSS)[14], Dynamic International Prognostic Scoring System (DIPSS)[15], or DIPSS-Plus prognostic scoring system[16] (Table 4). IPSS is suitable for newly diagnosed patients, while DIPSS and DIPSS-Plus are appropriate for prognostic assessment at any point during the patient’s course.
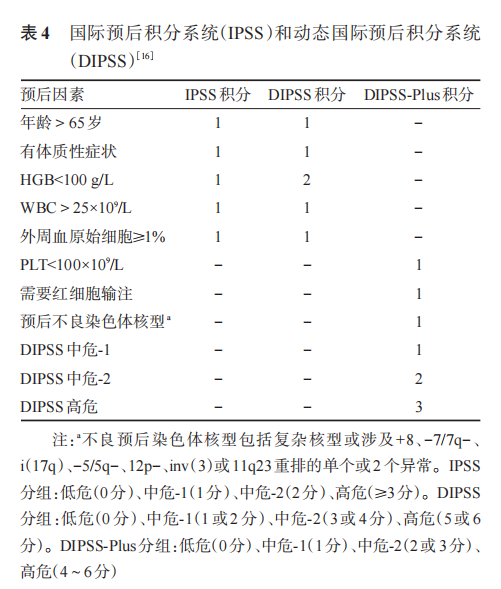
The IPSS (IPSS-Chinese) or DIPSS (DIPSS-Chinese) scoring revised for the characteristics of PMF in China is as follows: ① IPSS or DIPSS low-risk group (0 points); ② Intermediate risk-1, no palpable splenomegaly or PLT < 100×109/L (1 point); ③ IPSS or DIPSS intermediate risk-2 (2 points); ④ IPSS or DIPSS high risk (3 points). Based on the scores, patients are divided into low risk (0-1 points), intermediate risk (2-3 points), and high risk (4-5 points).
In recent years, with the elucidation of the mutation spectrum of PMF, the prognostic significance of gene mutations has also been preliminarily explored. An Italian research group combined JAK2, CALR, and MPL gene mutations with IPSS prognostic parameters to propose a new prognostic scoring system for PMF[18]: patients with constitutional symptoms (weight loss of 10% within one year before PMF diagnosis and/or unexplained fever or severe night sweats lasting more than one month), peripheral blood blast cells >1%, HGB <100g/L, JAK2V617F(+) each score 1 point; patients aged >65 years, WBC >25×109/L and MPL(+), without JAK2, CALR, and MPL gene mutations each score 2 points, classifying patients into very low risk (0 points), low risk (1 point), intermediate risk (2 or 3 points), high risk (4 points or 5 points), and very high risk (6 points or above). Studies have confirmed that this prognostic scoring system has a higher prognostic effect on patients than the IPSS system[19-20]. Recently, the MIPSS70 and MIPSS70-plus prognostic scoring systems[21] have also been proposed, but their clinical practical application value still needs further verification.
IV. Treatment
The treatment strategy for PMF can be formulated based on the patient’s prognostic group. For low-risk and intermediate risk-1 patients according to IPSS/DIPSS/DIPSS-Plus, if there are no obvious clinical symptoms and no significant anemia (HGB <100g/L), no significant splenomegaly (palpable left edge >10cm), elevated white blood cell count (>25×109/L) or significantly elevated platelet count (>1000×109/L), observation and monitoring of disease progression may be sufficient. If there are indications for cytoreductive therapy, hydroxyurea is the first choice, and IFN-α is also an effective cytoreductive drug[8].
Since PMF patients face a series of clinical issues such as anemia, splenomegaly, constitutional symptoms, and symptomatic extramedullary hematopoiesis, the current treatment strategies for PMF are primarily determined based on whether the patient has the aforementioned clinical problems, combined with the patient’s prognostic group for appropriate management[6-8].
(1) How to Treat Anemia
Treatment for anemia should begin when hemoglobin levels fall below 100g/L. Currently, drugs that have been proven effective for anemia in PMF include glucocorticoids, androgens, EPO, and immunomodulators, but all these drugs have shortcomings, and randomized controlled clinical trials are still lacking.
Androgens can improve anemia symptoms in 1/3 to 1/2 of patients, and glucocorticoids can improve symptoms in 1/3 of patients with severe anemia or thrombocytopenia. Therefore, patients with anemia and/or thrombocytopenia may initially be treated with a combination of androgens (stanozolol 6mg/d or danazol 200mg every 8 hours) and glucocorticoids (prednisone 30mg/d) for at least 3 months. If effective, the androgen can be continued, and the glucocorticoid can be gradually reduced. Androgens are not recommended for patients with prostate disease or liver disease.
The efficacy of EPO in treating PMF anemia is not uniform; some researchers have conducted meta-analyses of published literature, showing an efficacy rate of 30%-40% for EPO in treating PMF anemia. EPO is mainly suitable for anemic patients with serum EPO <100U/L, with a common dosage of 30,000-50,000U per week.
Conventional doses (>100mg/d) of thalidomide monotherapy have low efficacy and significant adverse reactions, and are not recommended as monotherapy. Low-dose thalidomide (50mg/d) combined with prednisone (0.5mg·kg-1·d-1) improves efficacy and reduces adverse reactions compared to thalidomide alone. Further improvement in efficacy and prolongation of effective duration can be achieved by adding danazol to the low-dose thalidomide and prednisone regimen[22]. Patients with grade 2 or higher peripheral neuropathy should not use thalidomide.
Results from phase II clinical trials of lenalidomide monotherapy for MF show that the efficacy rates for anemia, splenomegaly, and thrombocytopenia are 22%, 33%, and 50%, respectively. Lenalidomide (starting dose of 5mg/d for patients with PLT <100×109/L, and 10mg/d for patients with PLT ≥100×109/L, taken continuously for 21 days followed by 7 days off, with 28 days as one cycle) combined with prednisone (30mg/d) shows efficacy rates of 30% and 42% for anemia and splenomegaly, respectively.
(2) How to Treat Splenomegaly
Ruxolitinib can be used as first-line treatment for intermediate risk-2 and high-risk patients with splenomegaly according to IPSS/DIPSS/DIPSS-Plus, and can also be considered as first-line treatment for intermediate risk-1 patients with severe symptomatic splenomegaly (such as left upper abdominal pain or early satiety affecting food intake). For other patients, hydroxyurea is the preferred drug[8]. Radiation therapy in the splenic area only provides temporary benefits. Splenectomy remains a feasible option for patients with splenomegaly who do not respond to drug therapy.
1. Ruxolitinib: Two large phase III clinical trials, COMFORT-1 and COMFORT-2, have confirmed the efficacy of ruxolitinib in splenic reduction and improving symptoms related to bone marrow fibrosis, and have demonstrated that ruxolitinib significantly prolongs overall survival (OS) compared to existing conventional treatment drugs for bone marrow fibrosis. Five-year follow-up data from the COMFORT-1 and COMFORT-2 studies indicate that the mortality rate in the ruxolitinib treatment group is reduced by 30% compared to the control group, with median OS time extending from 45.9 months in the control group to 63.5 months in the ruxolitinib group. Additionally, the COMFORT-1 trial found that 33% of patients had improvements in the degree of bone marrow fibrosis, and 49% of patients remained stable, meaning that 82% of patients undergoing ruxolitinib treatment will stop or even improve the progression of bone marrow fibrosis. Results from international multicenter phase II clinical trials in mainland China (63 cases), Korea, Japan, and Taiwan are similar to those of COMFORT-1 and COMFORT-2[23].
The starting dose of ruxolitinib is primarily based on the patient’s platelet count level: the recommended starting dose for patients with pre-treatment PLT >200×109/L is 20mg twice daily; for patients with PLT (100~200)×109/L, the recommended starting dose is 15mg twice daily; for patients with PLT (50~<100)×109/L, the recommended starting dose is 5mg twice daily. The dose should not be increased during the first four weeks, and dose adjustments should be made at least two weeks apart, with a maximum dose of 25mg twice daily. If PLT <100×109/L during treatment, dose reduction should be considered; if PLT <50×109/L or absolute neutrophil count <0.5×109/L, treatment should be discontinued. Routine blood tests and detailed metabolic indicators, including uric acid and lactate dehydrogenase, should be checked before starting ruxolitinib treatment, and thereafter every 2-4 weeks until the ruxolitinib dose is stable, after which the frequency of follow-ups can be determined based on clinical conditions. The clinical symptom burden of the patient should also be assessed using MPN-10 before and during treatment. Additionally, monitoring of spleen size changes should be performed through palpation or ultrasound. Discontinuation should be gradual within 7-10 days, and sudden discontinuation should be avoided, with a recommendation to add prednisone 20-30mg/d during the discontinuation period.
The most common hematological adverse reactions of ruxolitinib are grade 3/4 anemia, thrombocytopenia, and neutropenia[24]. Grade 3/4 anemia can be seen in the first 6 months of treatment, primarily occurring in the first 8-12 weeks of treatment, reaching a steady state level around 24 weeks. For patients experiencing anemia during treatment, in addition to red blood cell transfusions, EPO or danazol can be added. Thrombocytopenia is the most common hematological adverse reaction within 8-12 weeks of treatment initiation, followed by a steady state of platelet counts. The primary management of thrombocytopenia is to adjust the ruxolitinib dosage based on platelet count levels.
The most common non-hematological adverse reactions of ruxolitinib are infections (especially urinary tract infections and respiratory infections) and viral reactivation[24]. A thorough inquiry into the patient’s past infection history (especially herpes zoster, tuberculosis, and hepatitis virus infection history) should be conducted before medication, along with routine screening for HIV and hepatitis viruses. Hepatitis virus carriers should have their viral load monitored dynamically during treatment.
2. Hydroxyurea: The efficacy rate for splenic reduction is about 40%. Patients who do not respond to hydroxyurea treatment can be switched to other myelosuppressive agents, such as intravenous cladribine (5mg·m-2·d-1×5d, with each infusion taking 2 hours, one cycle per month, repeated for 4-6 months), oral mercaptopurine (2.5mg three times a week), or oral busulfan (2-6mg/d, with close monitoring of blood counts).
3. Radiation Therapy in the Splenic Area: Radiation therapy can relieve symptoms of fullness caused by liver and splenic enlargement, but the symptom relief duration is short (median time 3-6 months). The total dose of radiation therapy in the splenic area is 0.1-0.5Gy (divided into 5-10 sessions). The main adverse reaction is cytopenia, with mortality rates from this cause exceeding 10%.
(3) How to Treat Constitutional Symptoms
Current assumptions suggest that the abnormal production of cytokines is causally related to the constitutional symptoms and cachexia associated with PMF. Constitutional symptoms in PMF patients can be severe and should be regarded as an important treatment indication. Treatment for splenomegaly often partially alleviates constitutional symptoms. Ruxolitinib can significantly improve constitutional symptoms in PMF; for those with MPN-10 total scores >44 or difficult and severe symptoms (individual scores >6) such as skin itching or unexplained weight loss (more than 10% in the past 6 months) or unexplained fever, ruxolitinib can be used as first-line treatment[25].
(4) How to Treat Extramedullary Hematopoiesis (EMH)
The thoracic vertebrae are the most common sites for PMF-related non-hepatic splenic extramedullary hematopoiesis (EMH). Other sites include lymph nodes, lungs, pleura, small intestine, peritoneum, urogenital tract, and heart. When clinical symptoms occur, low-dose localized radiation therapy (0.1-1.0Gy, divided into 5-10 sessions) can be applied. Currently, low-dose radiation therapy is a treatment option for PMF-related non-hepatic splenic EMH.
(5) Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT)[26-27]
allo-HSCT is currently the only treatment method that may cure PMF, but it carries a relatively high treatment-related mortality rate and complication incidence. The one-year treatment-related mortality rate for conventional intensity preconditioning allo-HSCT is about 30%, with an overall survival (OS) rate of about 50%; for reduced intensity preconditioning, the five-year median OS rate is about 45%, with treatment-related and relapse-related mortality rates being similar. In comparison, a recent study showed that PMF patients who met transplant criteria (high risk or intermediate risk-1 patients, <60 years old) but did not undergo HSCT had one-year and three-year OS rates ranging from 71%-95% and 55%-77%, respectively.
For patients with an estimated survival time of less than 5 years who meet the criteria for hematopoietic stem cell transplantation, the risks of allo-HSCT-related complications should be weighed. Candidates for allo-HSCT include patients with IPSS high risk (median OS period of 27 months) or intermediate risk-2 (median OS period of 48 months), as well as those who are transfusion-dependent (median OS period of 20 months) or have adverse cytogenetic abnormalities (median OS period of 40 months). Whether to ultimately choose allo-HSCT must also consider other adverse factors that could lead to allo-HSCT failure, including red blood cell transfusion load, severe splenomegaly, non-HLA matched sibling donors, high hematopoietic stem cell transplant comorbidity index (HCT-CI) scores, advanced age, late-stage disease, and non-HLA fully matched unrelated donors. If allo-HSCT is chosen, consultation with physicians experienced in hematopoietic stem cell transplantation is recommended.
(6) Splenectomy
The perioperative mortality rate for splenectomy in PMF is 5%-10%, with a postoperative complication rate of about 50%. Complications include surgical site bleeding, thrombosis, subphrenic abscess, accelerated liver enlargement, extreme platelet elevation, and leukocytosis with excessive blast cells. Patients considered for splenectomy must be in good physical condition and show no clinical or laboratory evidence of disseminated intravascular coagulation (DIC).
Indications for splenectomy include symptomatic portal hypertension (such as variceal bleeding, ascites), significantly symptomatic splenomegaly that is resistant to medication and accompanied by pain or severe cachexia, and transfusion-dependent anemia. In contrast, severe thrombocytopenia is a marker of impending leukemic transformation, and splenectomy is unlikely to improve prognosis in such patients. Recommended preventive measures before splenectomy include cytoreductive drugs and anticoagulants. Platelet counts should be maintained below 400×109/L, as extreme platelet elevations may occur postoperatively, and surgery should be performed by an experienced surgical team.
(7) Treatment in the Acute Transformation Phase[28]
Any treatment during this phase has poor efficacy, and experimental or palliative treatment should be considered. Intensive induction chemotherapy should be considered for selected patients, followed by allo-HSCT for consolidation. For patients planned for allo-HSCT, reversal of the disease to the chronic phase may not require complete remission.
V. Efficacy Assessment Criteria
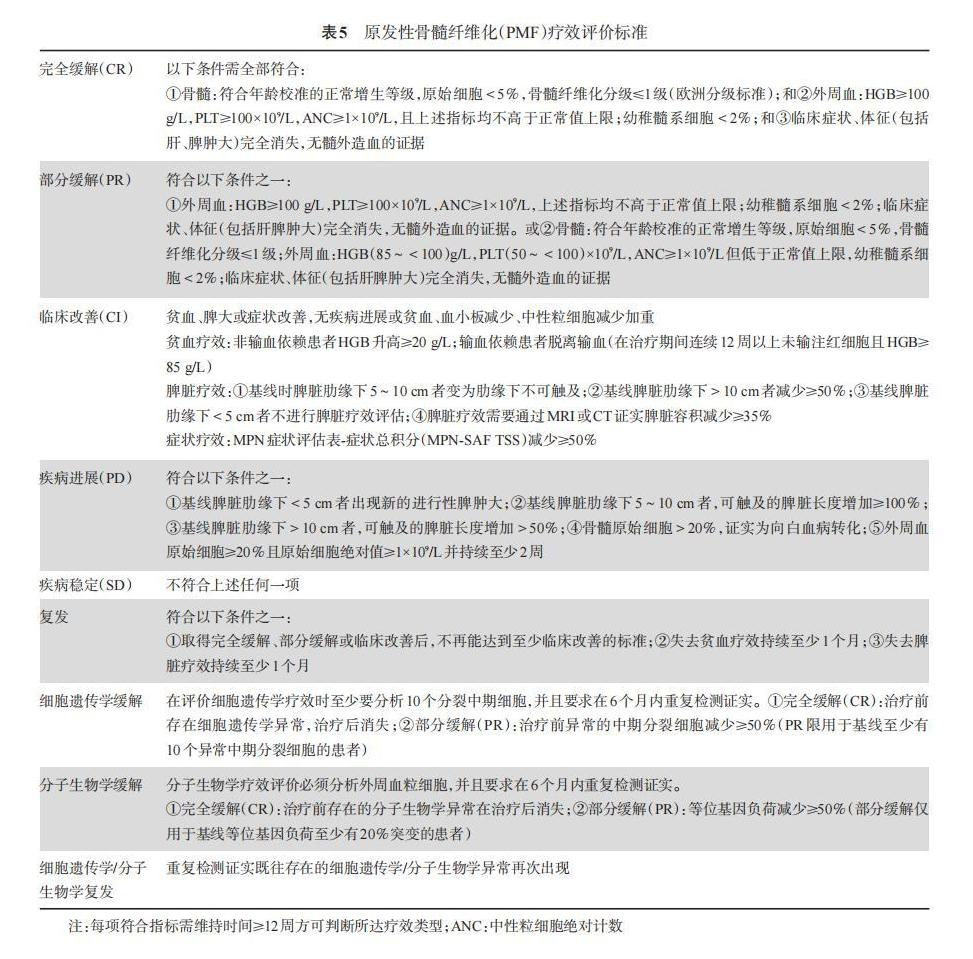
The efficacy assessment criteria proposed by the European Myelofibrosis Network (EUMNET) in 2005 evaluate efficacy from three levels: clinical hematological efficacy, bone marrow histological efficacy, and cytogenetic efficacy. In 2006, the International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) proposed an efficacy assessment standard to address the shortcomings of the previous criteria[29]. This guideline adopts the 2013 EUMNET and IWG-MRT consensus standards for efficacy assessment[30] (Table 5).
Experts participating in the guideline discussion (arranged by surname stroke order):
Chinese Academy of Medical Sciences Hematology Hospital (Wang Jianxiang, Xiao Zhijian, Zhang Peihong); Harbin Blood Oncology Research Institute (Ma Jun); Nanfang Hospital of Southern Medical University (Liu Qifa); West China Hospital of Sichuan University (Liu Ting, Pan Ling); Ruijin Hospital affiliated to Shanghai Second Medical University (Li Junmin); First Affiliated Hospital of Soochow University (Wu Depai, Chen Suning); First Affiliated Hospital of Nanjing University (Li Jianyong); Guangdong Provincial People’s Hospital (Du Xin); Peking Union Medical College Hospital (Zhou Daobin, Duan Minghui); Tianjin Medical University General Hospital (Shao Zonghong); Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (Hu Yu); Affiliated Union Hospital of Fujian Medical University (Hu Jianda); First Affiliated Hospital of Zhejiang University School of Medicine (Jin Jie); Shengjing Hospital of China Medical University (Yang Wei); Peking University People’s Hospital (Huang Xiaojun, Liu Kaiyan, Jiang Qian); Sixth People’s Hospital of Shanghai (Chang Chunkang)
References
[1] Chinese Medical Association Hematology Branch Leukemia and Lymphoma Group. Expert Consensus on the Diagnosis and Treatment of Primary Myelofibrosis in China (2015) [J]. Chinese Journal of Hematology, 2015, 36(9):721-725. DOI:10.3760/cma.j.issn.0253-2727.2015.09.001.
[2] Xu Junqing, Xu Zefeng, Wang Jingya, et al. Symptom burden assessment of 615 cases of Ph chromosome/BCR-ABL fusion gene negative myeloproliferative neoplasm patients [J]. Chinese Journal of Hematology, 2016, 37(1):26-29. DOI:10.3760/cma.j.issn.0253-2727.2016.01.005.
[3] Xiao Zhijian. Myeloproliferative neoplasms and myelodysplastic syndromes: Opening a new era of molecular diagnosis [J]. Chinese Journal of Hematology, 2014, 35(5):385-386. DOI:10.3760/cma.j.issn.0253-2727.2014.05.001.
[4] Bench AJ, White HE, Foroni L, et al. Molecular diagnosis of the myeloproliferative neoplasms: UK guidelines for the detection of JAK2V617F and other relevant mutations [J]. Br J Haematol, 2013, 160(1):25-34. DOI:10.1111/bjh.12075.
[5] Guglielmelli P, Pietra D, Pane F, et al. Recommendations for molecular testing in classical Ph1-negative myeloproliferative disorders: a consensus project of the Italian Society of Hematology [J]. Leuk Res, 2017, 58:63-72. DOI:10.1016/j.leukres.2017.04.006.
[6] Vannucchi AM, Barbui T, Cervantes F, et al. Philadelphia chromosome negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up [J]. Ann Oncol, 2015, 26 Suppl 5:v85-99. DOI:10.1093/annonc/mdv203.
[7] Mesa RA, Jamieson C, Bhatia R, et al. NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2.2018 [J]. J Natl Compr Canc Netw, 2017, 15(10):1193-1207. DOI:10.6004/jnccn.2017.0157.
[8] Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet [J]. Leukemia, 2018, 32(5):1057-1069. DOI:10.1038/s41375-018-0077-1.
[9] Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity [J]. Haematologica, 2005, 90(8):1128-1132.
[10] Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia [J]. Blood, 2016, 127(20):2391-2405. DOI:10.1182/blood-2016-03-643544.
[11] Buhr T, Hebeda K, Kaloutsi V, et al. European Bone Marrow Working Group trial on reproducibility of World Health Organization criteria to discriminate essential thrombocythemia from prefibrotic primary myelofibrosis [J]. Haematologica, 2012, 97(3):360-365. DOI:10.3324/haematol.2011.047811.
[12] Barbui T, Thiele J, Vannucchi AM, et al. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia [J]. Leukemia, 2013, 27(10):1953-1958. DOI:10.1038/leu.2013.74.
[13] Su Tao, Zhang Peihong, Xu Zefeng, et al. Clinical characteristics and prognosis analysis of primary myelodysplastic syndromes with bone marrow fibrosis [J]. Chinese Journal of Hematology, 2012, 33(5):378-382. DOI:10.3760/cma.j.issn.0253-2727.2012.05.007.
[14] Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment [J]. Blood, 2009, 113(13):2895-2901. DOI:10.1182/blood-2008-07-170449.
[15] Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWGMRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment) [J]. Blood, 2010, 115(9):1703-1708. DOI:10.1182/blood-2009-09-245837.
[16] Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status [J]. J Clin Oncol, 2011, 29(4):392-397. DOI:10.1200/JCO.2010.32.2446.
[17] Xu Z, Gale RP, Zhang Y, et al. Unique features of primary myelofibrosis in Chinese [J]. Blood, 2012, 119(11):2469-2473. DOI:10.1182/blood-2011-11-389866.
[18] Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis [J]. Blood, 2014, 124(7):1062-1069. DOI:10.1182/blood-2014-05-578435.
[19] Li B, Xu J, Wang J, et al. Calreticulin mutations in Chinese with primary myelofibrosis [J]. Haematologica, 2014, 99(11):1697-1700. DOI:10.3324/haematol.2014.109249.
[20] Li B, Zhang P, Feng G, et al. Bone marrow fibrosis grade is an independent risk factor for overall survival in patients with primary myelofibrosis [J]. Blood Cancer J, 2016, 6(12):e505. DOI:10.1038/bcj.2016.116.
[21] Guglielmelli P, Lasho TL, Rotunno G, et al. MIPSS70: mutation-enhanced international prognostic scoring system for transplantation-age patients with primary myelofibrosis [J]. J Clin Oncol, 2018, 36(4):310-318. DOI:10.1200/JCO.2017.76.4886.
[22] Luo X, Xu Z, Li B, et al. Thalidomide plus prednisone with or without danazol therapy in myelofibrosis: a retrospective analysis of incidence and durability of anemia response[J]. Blood Cancer J, 2018, 8(1):9. DOI: 10.1038/s41408-017-0029-4.
[23] Jin Jie, Du Xin, Zhou Daobin, et al. Efficacy and safety of JAK inhibitor ruxolitinib in Chinese patients with myelofibrosis: A2202 one-year follow-up results[J]. Chinese Journal of Hematology, 2016, 37(10): 858-863. DOI: 10.3760/cma.j.issn.0253-2727.2016.10.007.
[24] Saeed I, McLornan D, Harrison CN. Managing side effects of JAK inhibitors for myelofibrosis in clinical practice[J]. Expert Rev Hematol, 2017, 10 (7):617-625. DOI: 10.1080/17474086.2017.1337507.
[25] Marchetti M, Barosi G, Cervantes F, et al. Which patients with myelofibrosis should receive ruxolitinib therapy? ELN-SIE evidence-based recommendations[J]. Leukemia, 2017, 31(4):882-888. DOI: 10.1038/leu.2016.283.
[26] Kröger N, Giorgino T, Scott BL, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis[J]. Blood, 2015, 125(21):3347-3350. DOI: 10.1182/blood-2014-10-608315.
[27] Kröger NM, Deeg JH, Olavarria E, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group[J]. Leukemia, 2015, 29(11):2126-2133. DOI:10.1038/leu.2015.233
[28] Mascarenhas J, Heaney ML, Najfeld V, et al. Proposed criteria for response assessment in patients treated in clinical trials for myeloproliferative neoplasms in blast phase (MPN-BP): formal recommendations from the post-myeloproliferative neoplasm acute myeloid leukemia consortium[J]. Leuk Res, 2012, 36(12):1500-1504. DOI: 10.1016/j.leukres.2012.08.013.
[29] Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT)[J]. Blood, 2006, 108(5):1497-1503. DOI: 10.1182/blood-2006-03-009746.
[30] Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report[J]. Blood, 2013, 122(8):1395-1398. DOI: 10.1182/blood-2013-03-488098.


Scan to join the MPN patient group
Consult and communicate regarding issues related to myeloproliferative neoplasms (MPN)
Click to read the original text for more information


Click to share

Click to like

Click to view