
Chemotherapy, as one of the main options for cancer treatment, has limited selectivity for cancer cells, a narrow therapeutic window, and potential high toxicity, which restricts its clinical application. Antibody-drug conjugates (ADCs), as emerging anticancer therapeutics, can deliver highly cytotoxic molecules directly to cancer cells to exert their lethal effects.
ADCs are formed by the covalent bonding of monoclonal antibodies with cytotoxic chemical agents (payloads) through linkers. The linker plays a critical role in the therapeutic efficacy of ADCs, and its characteristics greatly influence the therapeutic index, pharmacodynamics, and pharmacokinetics of ADCs.
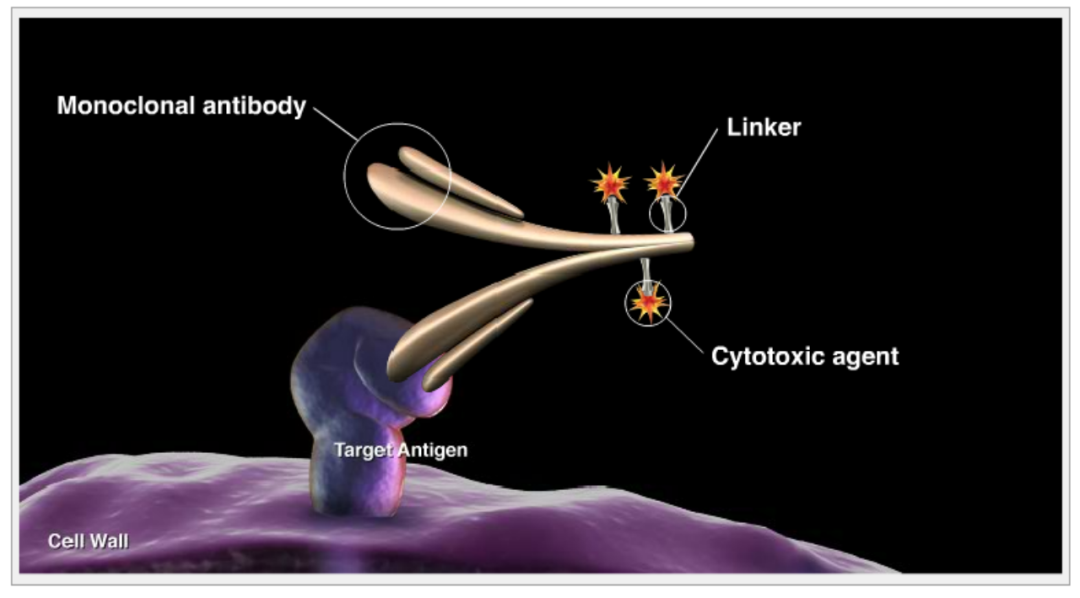
To ensure ADCs have selectivity and efficacy, the linkers used should strive to achieve three key characteristics[1]:
(1) High circulatory stability: The payload should not be released before reaching the target, thereby minimizing off-target effects;
(2) High solubility: This helps in conjugation and avoids the formation of inactive ADC aggregates;
(3) Effective release: Allows for the effective release of highly cytotoxic linker-payload metabolites.
Linker consists of two key components: antibody conjugation and Payload conjugation (Figure 1A)[2]. The bonding between the linker and mAb is crucial, as it determines the drug-to-antibody ratio (DAR), which in turn dictates the homogeneity and stability of the ADC; the bonding between the linker and payload is equally important.
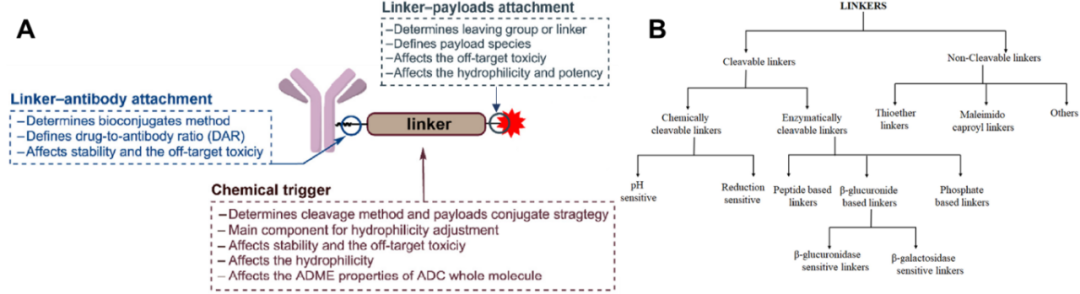
Figure 1. The role of linkers in ADCs (A) and their classification (B)
01
Differences, Advantages, and Disadvantages of Cleavable and Non-cleavable Linkers
The linkers used in ADCs are divided into cleavable and non-cleavable types (Figure 1B)[3]. For non-cleavable linkers, after entering the lysosome, the mAb is metabolized through a proteolytic mechanism, releasing metabolites that include the payload, linker, and amino acid moieties; if the key pharmacophore of the payload is not affected, significant modifications to the payload can still yield effective ADCs, as seen with Kadcyla®[4]. However, due to the lack of cell permeability of charged amino acid moieties, non-cleavable linkers often fail to exert bystander effects[5], thereby limiting the application range of ADCs containing non-cleavable linkers, which are mainly used for treating hematological cancers or tumors with high antigen expression.
In contrast to non-cleavable linkers, cleavable linkers release drugs under specific conditions at the target cells. Cleavable linkers can be further subdivided into chemically cleavable or enzymatically cleavable. Although cleavable linkers generally have a broader range of applicability compared to non-cleavable linkers, they exhibit greater instability in the bloodstream. Thus, the success of cleavable linkers depends on their ability to effectively distinguish between blood circulation conditions and target cell conditions.
Next, I will focus on various cleavable linkers used in the development of ADCs.
02
Chemically Induced Cleavable Linkers
There are three main types of chemically induced cleavable linkers: acid-cleavable, reducible (such as disulfides), and linkers that can be cleaved by external stimuli.
2.1 Acid-Cleavable Linker: Mylotarg®, Besponsa®, Trodelvy®
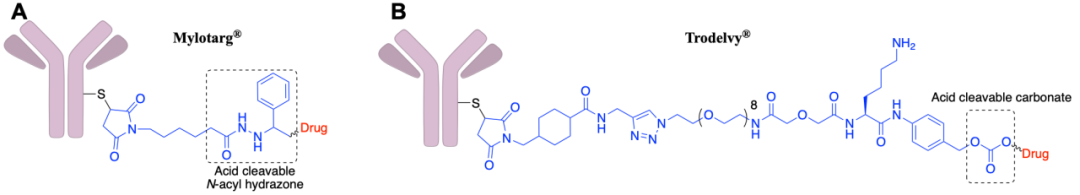
Acid-cleavable linkers are designed to take advantage of the acidity of endosomes (pH 5.5-6.2) and lysosomes (pH 4.5-5.0), while maintaining circulatory stability under physiological conditions at pH 7.4. This strategy achieved the earliest clinical success with Pfizer’s Mylotarg® (AcBut linker). While reducible disulfides were also employed, the linker includes acid-sensitive N-acylhydrazone bonds, which hydrolyze into ketones and acylhydrazones under acid catalysis (Figure 2A); during its development, a series of hydrazone linkers were tested for stability at pH 4.5 and pH 7.4, and those that were stable at pH 7.4 and unstable at pH 4.5 provided the most effective ADCs. This type of linker-payload has also been applied toBesponsa®.
In addition to the aforementioned hydrazone bonds, the carbonate linker used in Trodelvy® is also a type of acid-cleavable linker (Figure 2B). Although theoretically, ester bonds are more stable in blood circulation than carbonates, experimental results show that ADCs constructed with the former are not very stable in human serum; by introducing a p-aminobenzyl (PABC) spacer, the serum stability of the ADC can be significantly improved (t1/2=36 hrs), showing a certain selectivity for the acidic lysosomal compartment, with a t1/2 of 10 hours at pH 5.
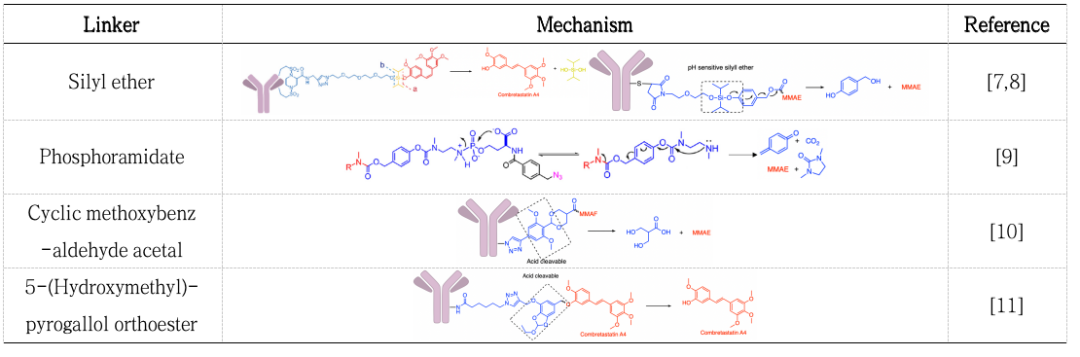
Besides the three marketed ADCs involving acid-cleavable linkers, numerous literature reports also discuss acid-cleavable (pH-sensitive) linkers (Table 1)[7-11].
Table 1. Mechanisms of Acid-Cleavable (pH-Sensitive) Linkers
2.2 Reducible Linkers
Despite the clinical successes of Mylotarg®, Besponsa®, and Trodelvy®, most ADC conjugation techniques no longer opt for acid-cleavable linkers. The requirement for linkers to strictly differentiate between pH 5 and pH 7.4 environments is inherently very challenging, and current research focuses on other methods that can yield higher tumor selectivity. While in some cases, slow release of the payload can yield beneficial results, this approach typically can only utilize payloads with moderate cytotoxicity; the currently preferred high-toxicity payloads require more stable linkers.
In Mylotarg® and Besponsa®, the release of the payload relies not only on the acid-sensitive hydrazone bonds but also on the disulfide bonds in the linker. The disulfide bonds are stable at physiological pH but are susceptible to nucleophilic attack by thiols (Figure 3A). In plasma, the main thiol species is the reduced form of human serum albumin (HAS, ~422 μM), but its reactivity towards macromolecules is hindered due to the limited solvent exposure of free thiol-containing residues; in contrast to the limited reducing capacity in plasma, the cytoplasm contains high levels of glutathione (GSH, 1–10 mM); the reducing conditions in plasma and cytoplasm provide opportunities for ADCs to selectively release payloads[12]. Moreover, compared to normal tissues, tumor-associated oxidative stress often leads to elevated GSH levels, increasing additional selectivity for cancer cells.
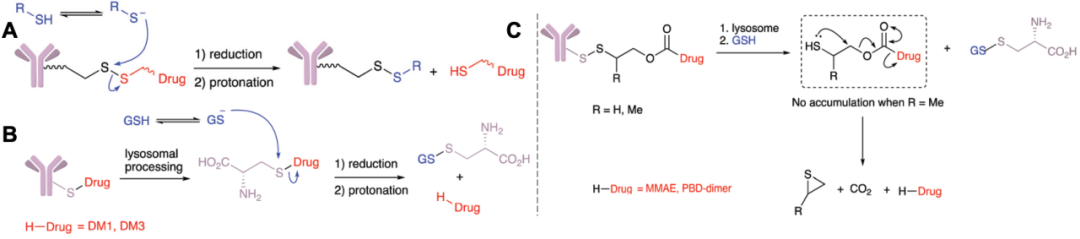
Figure 3. Hydrolysis mechanisms of ADC drugs under reducing conditions
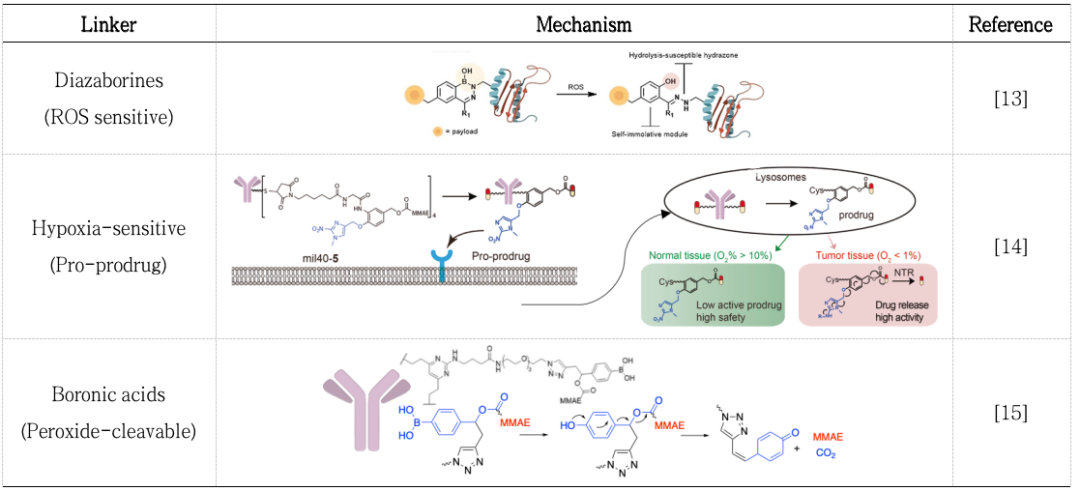
Linkers involving hydrolysis of disulfide bonds often contain maytansine derivatives (DM1/3/4, Figure 3B) and disulfide carbamate payloads (Figure 3C). Similar to disulfide bonds, payloads can be released under reducing conditions; numerous literature reports have indicated that linkers can release payloads in the highly reducing conditions of tumor tissues (Table 2)[13-15].
Table 2. Mechanisms of Linker Cleavage under Reducing Conditions
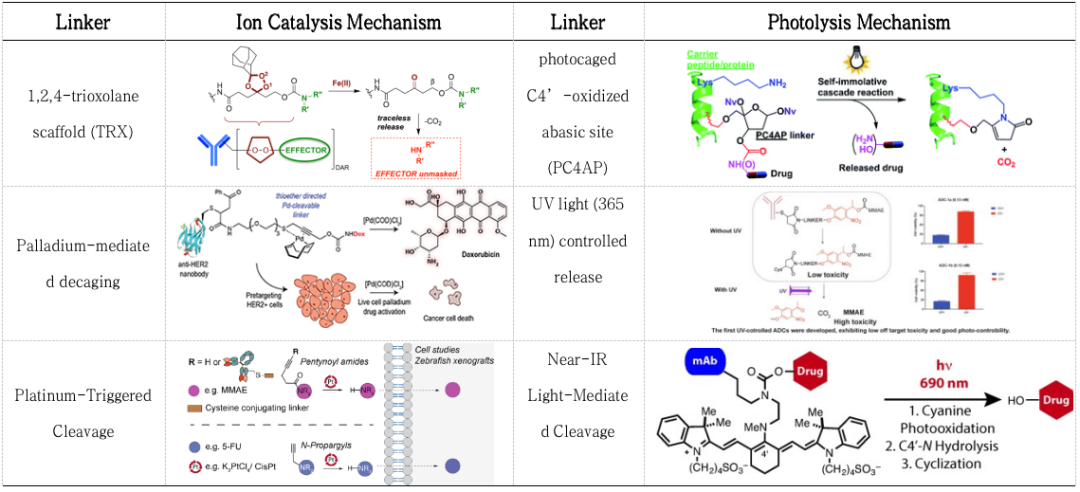
Although utilizing endogenous cleavage of ADC linkers is the simplest method for drug release, releasing payloads through external stimuli can have the following advantages: (1) avoiding differences in linker cleavage rates due to biological variations among patients, and (2) allowing ADCs to function when endogenous cleavage is insufficient to effectively release payloads. The cleaving mechanism used in Akalux®, which has been marketed in Japan, is a near-infrared light-sensitive linker that releases the toxic IRDye700 upon exposure to 690 nm red light, belonging to the category of near-infrared immunotherapy. Additionally, there is ongoing research on metal ion (Pt, Pd, Fe, Lu) catalyzed cleavage, UV/Vis, and NIR light-sensitive linkers (Table 3)[16-22].
Table 3. Linker Cleavage Induced by External Stimuli
03
Enzyme-Mediated Linker Cleavage
In the classic mechanism of ADC action, ADCs are transported to the lysosomes of cells, where high concentrations of unique hydrolytic enzymes are present, providing opportunities for enzyme-cleavable linkers to selectively cleave within cells. Currently, the application of cathepsin B is the most successful, with marketed drugs including Adcetris®, Polivy®, Padcev®, Tivdak®, Aidixi®, Lumoxiti®, Zynlonta®, Enhertu® all utilizing cathepsin B-cleaved peptide linkers, with p-aminobenzyl carbamate (PABC) used as a self-degradable spacer that spontaneously undergoes 1,6-elimination after proteolysis, releasing payloads, CO2, and nitrogen-containing quinone methylation products (Figure 4); PABC maintains enzyme activity independent of the payload, increasing the applicability of these peptide linkers. All these linker combinations exhibit certain stability in isolated human plasma.
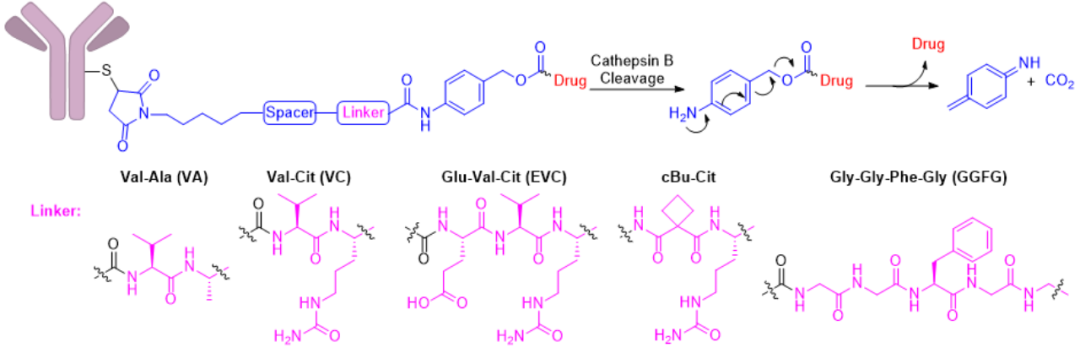 Figure 4. Structure and cleavage mechanism of peptide linkers
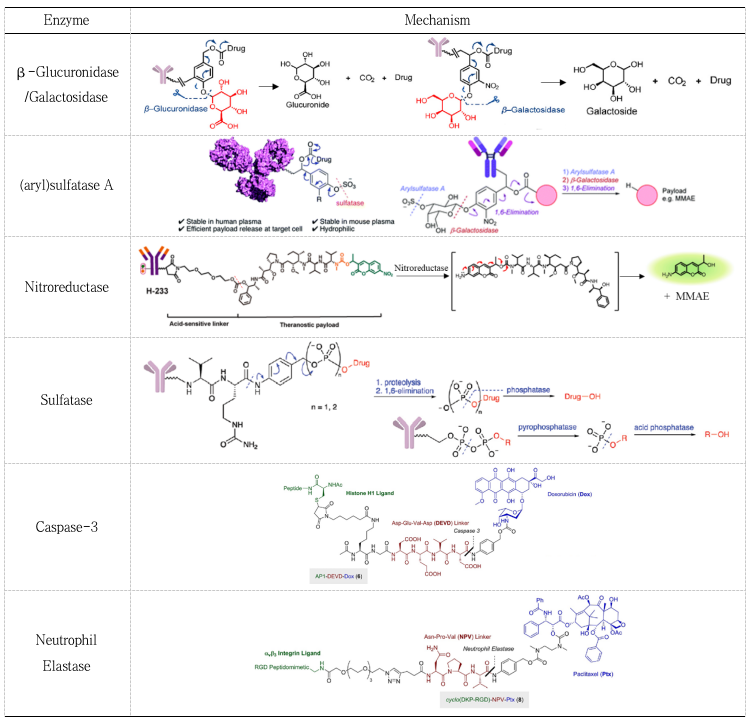
Figure 4. Structure and cleavage mechanism of peptide linkersIn addition to the commonly used enzyme cathepsin B, other enzymes such as phosphatases, (aryl) sulfatases, β-galactosidases, β-glucosidases, and nitroreductases have also been applied in the construction of ADCs (Table 4)[1-3, 23-28].
Table 4. Mechanisms of Enzyme-Mediated Linker Cleavage
04
Other Cleavable Linkers
Based on the principles of bioorthogonal chemistry, which does not interfere with normal biological processes, has high selectivity, is quick and simple to handle, and produces non-toxic by-products, bioorthogonal cleavage can also serve as a trigger for linker cleavage. Classic bioorthogonal cleavage using Cu(I)-2-[4-[[bis[(1-tert-butyltriazole-4-yl)methyl]amino]methyl]triazole-1-yl]acetic acid (BTTAA) and doubly substituted alkyne carbonyl (dsProc) reactions releases payloads on the surface of cancer cells (Figure 5A)[29]. In vitro toxicity experiments indicate that adding 50 mmol/L Cu(I)-BTTAA can reduce the IC50 of ADC containing DOX-dsProc by 120 times. Although this linker expands the cleavage mechanisms, improvements are still needed in substrate stability, biocompatibility, and operational convenience, and it is not yet applicable in vivo, indicating a long way to go before clinical application.
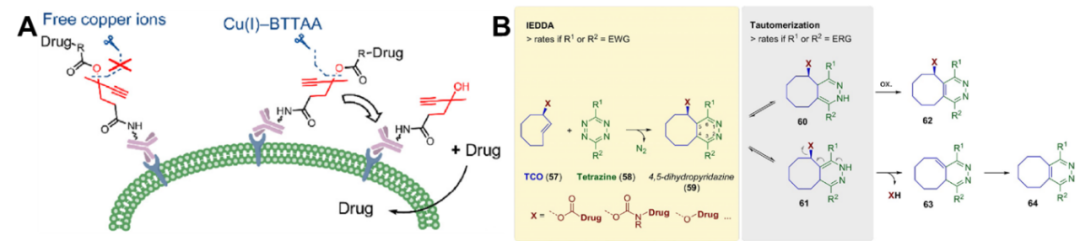
In addition to the aforementioned reactions, the IEDDA reaction can also be used for payload release. This method, introduced by Tagworks Pharmaceuticals, utilizes trans-cyclooctene (TCO) as a cleavable linker, which undergoes a click reaction with tetrazine activators to form a 4,5-dihydrodiazine intermediate, which is then converted into 2,5- and 1,4-isomers, of which only the latter can undergo subsequent electron cascade reactions to release different payloads (Figure 5B)[30]. The IEDDA and tautomerization steps are influenced by the substituents on the tetrazine, and a combination of two functional groups with opposing electron-withdrawing and electron-donating properties shows better drug release performance; additionally, acid catalysis can accelerate the release rate of payloads by affecting the tautomerization following the click reaction. Recent preclinical experiments have demonstrated the feasibility of the “click and release” method using the tetrazine-TCO pair[22].
Outlook
Future research on cleavable linkers is expected to further explore the possibilities of using multiple mechanisms of action, including the field of externally induced cleavage, which is still in its infancy. Although preclinical results of non-peptide enzyme-cleavable linkers are encouraging, they have yet to be clinically validated. The development of new cleavable linkers faces significant challenges, but as we gain deeper insights into the effects of different payloads on tumor biology and the clinical validation of optimal linker-spacer connections, we will ultimately develop ideal ADCs with disruptive efficacy to advance personalized medicine to meet the urgent medical needs in oncology.
 Main References
Main References