Introduction
The Zeng Lab’s public account aims to share cutting-edge information on the supramolecular self-assembly and excited-state properties of naphthalene diimide/perylene diimide (NDI/PDI), while also serving as a window to introduce the team’s work and share daily activities. Due to the limited level of the editor, there may inevitably be errors, and we welcome corrections from experts and scholars.
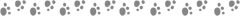
 Today, we share an article published in 2024 in the Journal of the American Chemical Society by Northwestern University, titled “Charge Transfer Dynamics in Supramolecular Tessellations Composed of Aromatic Donors and Chiral Tris(naphthalenediimide) Triangular Acceptors.” Understanding the charge transfer (CT) dynamics in donor-acceptor (D-A) co-crystals is crucial for developing efficient organic photovoltaic and electronic materials. The paper explores the photogenerated CT states in supramolecular chimeric structures formed by chiral tris(naphthalenediimide) triangular acceptor (−)-NDI-Δ co-crystallized with electron donors such as pyrene, perylene, and dibenzo[b,def]anthracene (PXX). By modulating crystallization conditions, one-dimensional (1D) and two-dimensional (2D) co-crystals with different structural features and morphologies were prepared. Using femtosecond and nanosecond transient absorption microscopy (fs-TAM and ns-TAM) and time-resolved electron paramagnetic resonance spectroscopy (TR-EPR), the dynamics of the CT states in co-crystals of different dimensions were elucidated. The study found that compared to 1D co-crystals, the lifetime of the CT state in 2D co-crystals is significantly extended, which can be attributed to the differences in structural symmetry and molecular packing modes affecting the CT interactions. This work highlights the potential of using pre-organized covalent multipoint charge carriers as donors/acceptors in co-crystals to construct multifunctional advanced materials with tunable CT characteristics.
Today, we share an article published in 2024 in the Journal of the American Chemical Society by Northwestern University, titled “Charge Transfer Dynamics in Supramolecular Tessellations Composed of Aromatic Donors and Chiral Tris(naphthalenediimide) Triangular Acceptors.” Understanding the charge transfer (CT) dynamics in donor-acceptor (D-A) co-crystals is crucial for developing efficient organic photovoltaic and electronic materials. The paper explores the photogenerated CT states in supramolecular chimeric structures formed by chiral tris(naphthalenediimide) triangular acceptor (−)-NDI-Δ co-crystallized with electron donors such as pyrene, perylene, and dibenzo[b,def]anthracene (PXX). By modulating crystallization conditions, one-dimensional (1D) and two-dimensional (2D) co-crystals with different structural features and morphologies were prepared. Using femtosecond and nanosecond transient absorption microscopy (fs-TAM and ns-TAM) and time-resolved electron paramagnetic resonance spectroscopy (TR-EPR), the dynamics of the CT states in co-crystals of different dimensions were elucidated. The study found that compared to 1D co-crystals, the lifetime of the CT state in 2D co-crystals is significantly extended, which can be attributed to the differences in structural symmetry and molecular packing modes affecting the CT interactions. This work highlights the potential of using pre-organized covalent multipoint charge carriers as donors/acceptors in co-crystals to construct multifunctional advanced materials with tunable CT characteristics.
Introduction
Organic D-A co-crystals have been proven useful for elucidating the formation and migration dynamics of charge transfer (CT) excitons in the solid state. Achieving efficient solar energy conversion in single D-A co-crystals requires meeting three core criteria: (1) rapid formation of CT excitons; (2) efficient generation of free charge carriers from CT excitons; (3) high charge mobility to effectively reach interfaces or electrodes. Two typical stacking structures are commonly found in D-A co-crystals: alternating …DADA… π-stacking and segregated …DDDD… and …AAAA… stacking. These stacking patterns arise from π-π interactions, CT interactions, and non-covalent forces such as halogen bonds and hydrogen bonds. The close packing between D and A leads to strong electronic coupling, often resulting in CT absorption bands in electronic spectra, allowing CT states to be directly photogenerated. However, this strong coupling can also lead to rapid charge recombination, inhibiting the diffusion and dissociation of CT excitons. Although some D-A co-crystals exhibit long-lived CT states with significant mobility, further extending their lifetimes remains a challenge in the field of crystal engineering. Therefore, there is an urgent need to introduce additional degrees of freedom to modulate the D-A interactions within the crystal. One effective strategy is to introduce pre-organized covalent multipoint charge carriers as donors or acceptors in the co-crystal to enhance CT characteristics.
Naphthalene diimide (NDI) is prone to undergo reversible single-electron and double-electron reduction reactions and is amenable to functional modification, making it an ideal module for studying electron transport, D-A interactions, and electron delocalization phenomena. The ordered supramolecular structures formed by the assembly of donor and acceptor molecules through non-covalent interactions—organic co-crystals—offer multiple advantages in electronics and photovoltaic applications: their preparation methods are similar to traditional single-component crystallization, making them simple and accessible; the electronic properties of the co-crystals can be customized by modulating molecular packing and morphology without additional chemical synthesis; they often exhibit emergent properties not possessed by the components themselves, providing possibilities for developing new functional materials. This work focuses on the supramolecular chimeric structures obtained from co-crystallization of chiral triangular prism (−)-NDI-Δ with electron donors pyrene (PYR), perylene (PER), and dibenzo[b,def]anthracene (PXX), studying their photogenerated CT states; (−)-NDI-Δ is formed by three electron-deficient NDI units bridged by (R,R)-trans-1,2-diaminocyclohexane, and has been used to explore electron transfer between NDI units, allowing for rapid electron hopping among the three NDI centers after single-electron reduction and capable of reversibly accepting up to six electrons; previous studies have shown that crystallization conditions can significantly modulate the single crystal and D-A co-crystal structures of (−)-NDI-Δ, endowing them with highly tunable optoelectronic properties. In this context, it is essential to consider: (1) the rigid triangular configuration of (−)-NDI-Δ can form various chimeric packing modes; (2) the triangular cavity can impose confinement on guest molecules to form host-guest complexes; (3) the intramolecular electron hopping among the three NDI units within (−)-NDI-Δ provides additional degrees of freedom for charge transport in the chimeric packing.
By modulating the characteristics of the crystallization solvent—such as its ability to occupy the cavity of (−)-NDI-Δ and participate in hydrogen bonding interactions—the authors selectively grew 1D or 2D co-crystals with distinct morphologies. Solid-state structural analysis indicated that the donor-acceptor π-π overlap is greater in 1D co-crystals, leading to stronger CT interactions. This series of co-crystals thus exhibits differences in the lifetimes of the CT states between 1D and 2D structures. Using femtosecond/nanosecond transient absorption microscopy and time-resolved electron paramagnetic resonance, the authors confirmed that this lifetime difference arises from differences in molecular packing symmetry. Overall, using pre-organized covalent multipoint charge carriers as donors or acceptors in co-crystals not only provides tunable supramolecular structures and photodriven behavior but also offers important insights into the structure-activity relationships between supramolecular structures and charge transfer characteristics.
Scheme 1. (−)-NDI-Δ with three electron donors: perylene (PER), dibenzo[b,def]anthracene (PXX), and pyrene (PYR).
Experimental Results
1: Co-crystal Design and Synthesis
Co-crystallizing the electron-deficient (−)-NDI-Δ with electron-rich aromatic donors PER, PXX, or PYR in a 1:1 molar ratio yields six types of CT co-crystals: (−)-NDI-Δ-PYR 1D, (−)-NDI-Δ-PYR 2D, (−)-NDI-Δ-PXX 1D, (−)-NDI-Δ-PXX 2D, (−)-NDI-Δ-PER 1D, and (−)-NDI-Δ-PER 2D, with specific morphologies depending on the crystallization conditions. Slowly diffusing a donor solvent such as MeCN into an ether solution containing (−)-NDI-Δ and the donor selectively yields needle-like 1D CT complexes, with a molar ratio of (−)-NDI-Δ:donor of 1:1 or 1:2, and one MeCN molecule occupying the cavity of (−)-NDI-Δ; all 1D co-crystals are needle-like, with (−)-NDI-Δ molecules stacked in a face-to-face slip arrangement. Slowly cooling a refluxing DMSO solution containing (−)-NDI-Δ and the donor selectively yields block-like 2D CT complexes, with a molar ratio of (−)-NDI-Δ:donor of 2:1, and (−)-NDI-Δ molecules arranged in alternating helical stacks, forming a hexagonal arrangement along the c-axis. Slowly cooling a 10:1 DMSO:hexane solution of (−)-NDI-Δ and PYR can simultaneously yield both 1D and 2D single crystals, with structures consistent with previous reports.
1D Co-crystal Structure of (−)-NDI-Δ-Donor
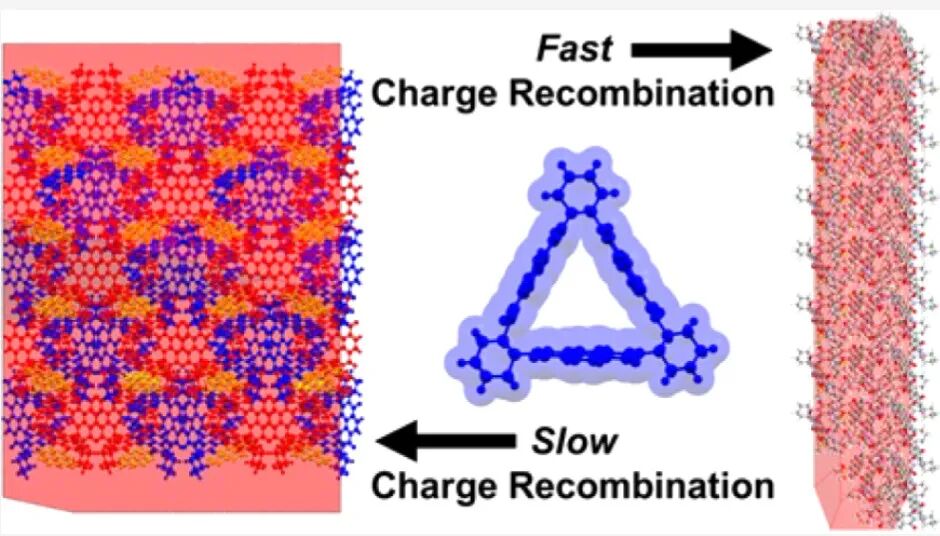
All 1D co-crystals of (−)-NDI-Δ with donors are needle-like, with hexane or MeCN molecules embedded in the cavity of (−)-NDI-Δ. These solvent molecules extend from the cavity and form hydrogen bonds with adjacent (−)-NDI-Δ, hindering its typical C3 rotation, resulting in edge-face (PYR system) or slip (PER, PXX systems) packing between molecules. Except for (−)-NDI-Δ-PXX 1D, various π-π distances are observed in the structure, attributed to the competition between solvent guests and π-π interactions, disrupting the classical chimeric arrangement of (−)-NDI-Δ, leading to alternating arrangements along the b or c axis.
Figure 1. (a) Observing the (−)-NDI-Δ-PYR 1D co-crystal superstructure along the b-axis; (b) observing the (−)-NDI-Δ-PXX 1D superstructure along the b-axis; (c) observing the (−)-NDI-Δ-PER 1D superstructure along the c-axis. (−)-NDI-Δ is red/blue, PYR is yellow, PXX is green, PER is yellow.
2D Co-crystal Structure of (−)-NDI-Δ-Donor
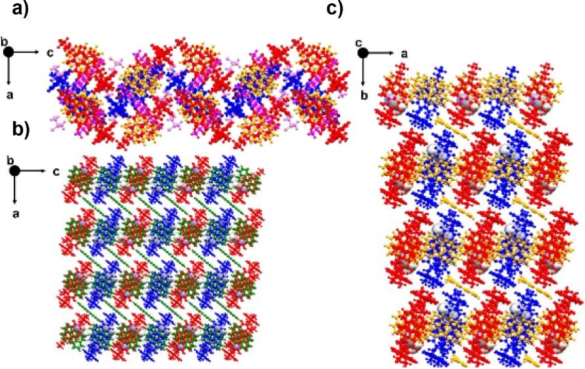
All 2D co-crystals of (−)-NDI-Δ-donor are block-like, with no solvent molecules in the cavity of (−)-NDI-Δ. The absence of solvent allows adjacent (−)-NDI-Δ molecules to exhibit classical C3 rotational hydrogen bonding interactions, forming cavities with a center-to-center distance of 7.91 Å and a lateral slip of 2.46 Å. In the 2D structures of (−)-NDI-Δ-PYR and (−)-NDI-Δ-PXX, the π-π stacking distances between the donor and (−)-NDI-Δ are uniform; however, (−)-NDI-Δ-PER 2D exhibits two different π-π distances. The 1D co-crystals show various π-π distances, attributed to solvent insertion disrupting the packing. The 2D co-crystals exhibit classical honeycomb chimeras along the c or a axis.
Figure 2. Observing the extended packing of the 2D crystal superstructure along the c-axis. (a) (−)-NDI-Δ-PYR 2D; (b) (−)-NDI-Δ-PXX 2D; (c) (−)-NDI-Δ-PER 2D. NDI-Δ is red/blue, DMSO solvent is yellow, PXX is green, PER is yellow.
2: Steady-State Absorption Spectra
(−)-NDI-Δ-PYR exhibits two strong peaks at 500 and 550 nm, significantly red-shifted compared to the monomer, due to the strong CT interaction between PYR and (−)-NDI-Δ, with 550 nm being the lowest CT transition. The absorption of the 1D needle-like co-crystal is sensitive to polarization (Figure 3a): the π-stacking is aligned along the needle’s long axis, and the transition dipole moment is parallel to it, resulting in a significant difference in intensity between parallel and perpendicular orientations. The 2D block-like co-crystal’s polar coordinate plot is nearly circular (Figure 3b,d,f), as the π-stacking plane is parallel to the illumination plane and isotropic, averaging the polarization dependence of the CT transition, exhibiting almost no polarization dependence.
(−)-NDI-Δ-PER co-crystal shows broad absorption bands at 525 and 675 nm, while (−)-NDI-Δ-PXX co-crystal exhibits similar bands at 500 and 610 nm, with both showing equal absorption points at 625 and 560 nm, indicating significant Davydov splitting; these bands are significantly red-shifted compared to the absorptions of PER, PXX, and (−)-NDI-Δ monomers, attributed to the strong CT transitions between the donor and (−)-NDI-Δ. In the PXX system, the transition dipole moment between the donor and (−)-NDI-Δ couples due to offset matching, resulting in discernible double peaks; in the PER system, the relative offset of the donor to NDI due to molecular size and sterics also leads to splitting. Despite the different donors causing variations in the absorption spectra, the polarization dependence of the 1D and 2D crystals still shows a consistent trend: in the 1D needle-like crystal, the π-stacking is aligned along the long axis, and the CT transition dipole moment is also along the long axis, exhibiting strong polarization correlation; in the 2D block-like crystal, the π-stacking plane is parallel to the light-illuminated surface and isotropic, averaging the polarization effect, resulting in a nearly circular shape with no polarization dependence. The CT state energy levels of all co-crystals are estimated from the longest wavelength absorption edge, with 1D and 2D being essentially consistent: approximately 2.1 eV for the PYR system, approximately 1.7 eV for the PER system, and approximately 1.6 eV for the PXX system.
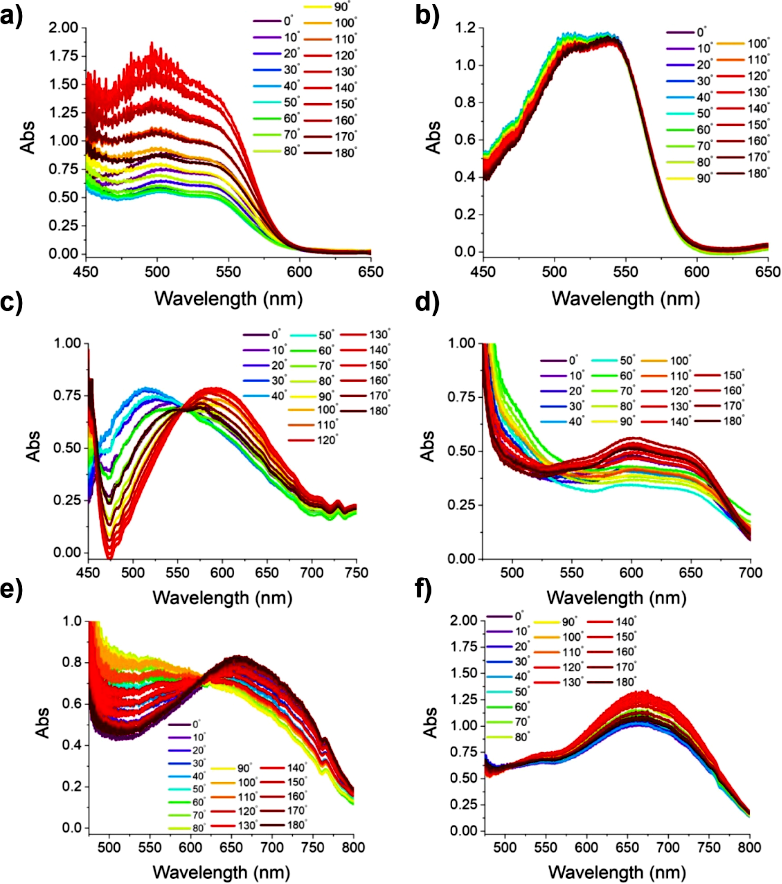
Figure 3. (a) (−)-NDI-Δ-PYR 1D, (b) (−)-NDI-Δ-PYR 2D, (c) (−)-NDI-Δ-PER 1D, (d) (−)-NDI-Δ-PER 2D, (e) (−)-NDI-Δ-PXX 1D, and (f) (−)-NDI-Δ-PXX 2D co-crystals’ polarization-resolved steady-state absorption spectra; the angles in the figure represent the polarization direction of linearly polarized light relative to the long axis of the 1D co-crystal.
3: Transient Absorption Microscopy
In all single crystal tests, a pump wavelength that has minimal effect on absorption (λexc) was selected for excitation. Exciting the (−)-NDI-Δ-PYR co-crystal at 550 nm results in a broad excited-state absorption (ESA) centered at 600 nm, attributed to NDI•–; PYR•+ is located at 453 nm but was not detected. The signal is generated within <300 fs and subsequently decays to a slightly red-shifted ESA at ~610 nm, with a lifetime exceeding 7 ns. Global fitting (A→B model) yields the evolution-associated spectra: A represents the PYR•+-NDI•– CT exciton, which transitions to a long-lived state at 1099 ± 4 ps and 2411 ± 19 ps for 1D and 2D co-crystals, respectively. ns-TAM shows that this state returns to the ground state through 1D annihilation, with rate constants of (1.70 ± 0.01) and (1.60 ± 0.01) ΔA-1 μs-1.
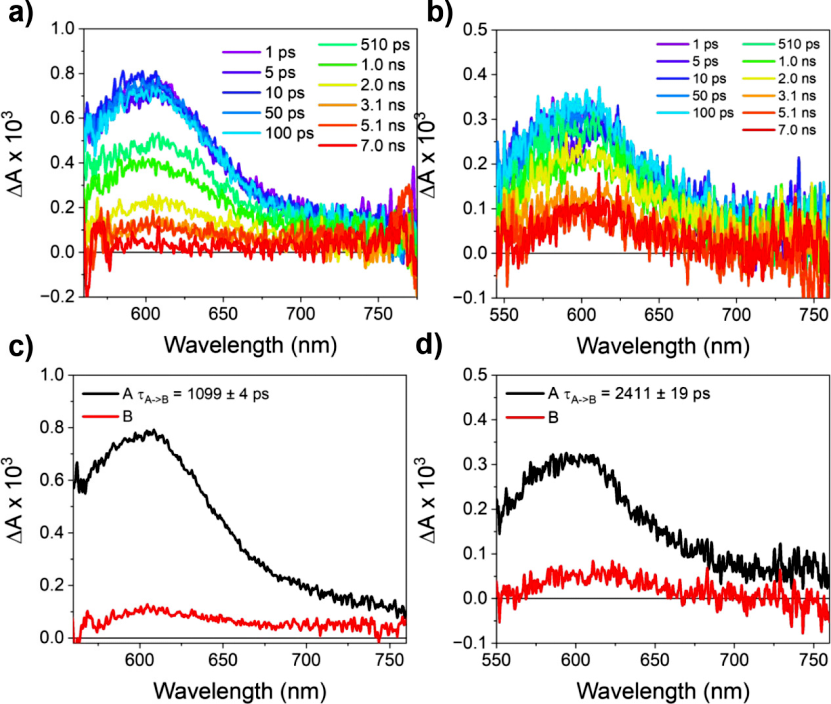
Figure 4. (−)-NDI-Δ-PYR 1D and 2D single crystal femtosecond transient absorption microscopy data. (a,b) fs-TAM spectra for 1D (λexc = 550 nm) and 2D (λexc = 550 nm) as a function of pump-probe delay. The probe light for the 1D co-crystal is polarized along the long axis, while for the 2D co-crystal, it is polarized along the direction of maximum absorption. (c,d) Evolution-associated spectra obtained using the A→B model.
It is not possible to determine the identity of the long-lived state solely from the TAM data: the 610 nm band resembles the ESA of NDI•– and the previously reported (NDI•–-PYR•+) CT triplet absorption spectrum, both of which can persist in single crystals. To clarify its attribution, we performed time-resolved EPR (TREPR) on powdered crystal samples (Figure 5). The simulated experimental TREPR spectra of (−)-NDI-Δ-PYR 1D and 2D indicate that the long-lived state is a spin-polarized triplet, with zero-field splitting parameters |D|/|E| of 698/117 MHz and 769/118 MHz, significantly smaller than the intrinsic values of 3*PYR and 3*NDI; and exhibiting an (e,e,e,a,a,a) polarization pattern, consistent with spin-orbit charge transfer intersystem crossing (SOCT-ISC), ruling out the mechanism of free radical intersystem crossing.
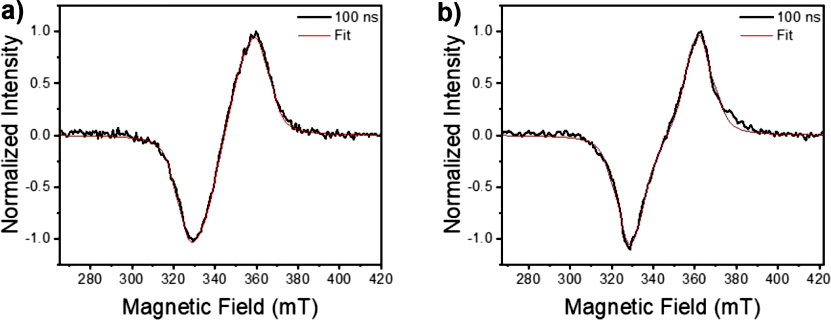
Figure 5. At 85 K, using 5 mW microwave power, TREPR spectra collected 100 nm after excitation with a 585 nm laser pulse: (a) (−)-NDI-Δ-PYR 1D co-crystal powder; (b) (−)-NDI-Δ-PYR 2D co-crystal powder. The red curve represents the data simulation. To reproduce these broad and featureless spectra, a larger Lorentzian line width (ΓL = 4 mT) is required.
The |D| value is reduced compared to the expected value for isolated chromophore triplets, typically attributed to charge transfer (CT) interactions with neighboring molecules, where the triplet CT state mixes with the localized neutral triplet on the chromophore. This increases the average distance between the two electrons constituting the triplet exciton, thereby reducing the electron-electron dipole interaction |D|. Using the point dipole approximation and taking |D| = 698 MHz and 769 MHz, the average distances between the two unpaired electrons in the triplet exciton are calculated to be 4.8 Å and 4.7 Å, respectively; while the actual distances between the donor and acceptor in the co-crystal structure are 3.34 Å and 3.40 Å. Therefore, the TREPR data indicate that the triplet exciton has a high CT character, consistent with our previous observations in NDI-PYR co-crystals. The time constant τA→B represents the formation time of the triplet, and its decay occurs through triplet-triplet annihilation (TTA). The rapid SOCT-ISC may arise from the charge delocalization in the initially formed 1CT state, which is constituted by a pair of face-to-face oxidized donors and a single reduced (−)-NDI-Δ. SOCT-ISC has also been observed in D-A molecules composed of BODIPY, perylene diimide, and carbazole, suggesting that this mechanism may be generally applicable as long as the triplet state is energetically accessible. However, ISC may also be more complex, mediated by vibrational coupling between 3CT and localized exciton triplet states (3LE), thereby enhancing the spin-orbit coupling of the 1CT state, as reported in thermally activated delayed fluorescence (TADF) systems. The long lifetime of the triplet further increases its chances of encountering in the lattice, leading to efficient annihilation in the solid state.
Exciting the (−)-NDI-Δ-PER 1D and 2D co-crystals with 690 nm generates broad ESA centered at 560 nm and 775 nm, which also features (−)-NDI-Δ•– (Figure 6). In solution, PER•+ has an absorption peak at approximately 537 nm, overlapping with the broad and strong ESA of (−)-NDI-Δ•–. These absorption changes occur within <300 fs and rapidly return to the ground state at 140.0 ± 0.2 ps (1D) and 245.0 ± 0.3 ps (2D). Exciting the (−)-NDI-Δ-PXX single-component co-crystal at 780 nm yields similar results (Figure 7). Again, broad ESA centered at 575, 700, and 800 nm is observed, featuring (−)-NDI-Δ•–. Additionally, a strong ESA appears around 940 nm, attributed to PXX•+. These absorption changes also occur within <300 fs and rapidly decay back to the ground state at τ = 17.0 ± 0.2 ps (1D) and 37.0 ± 0.3 ps (2D).
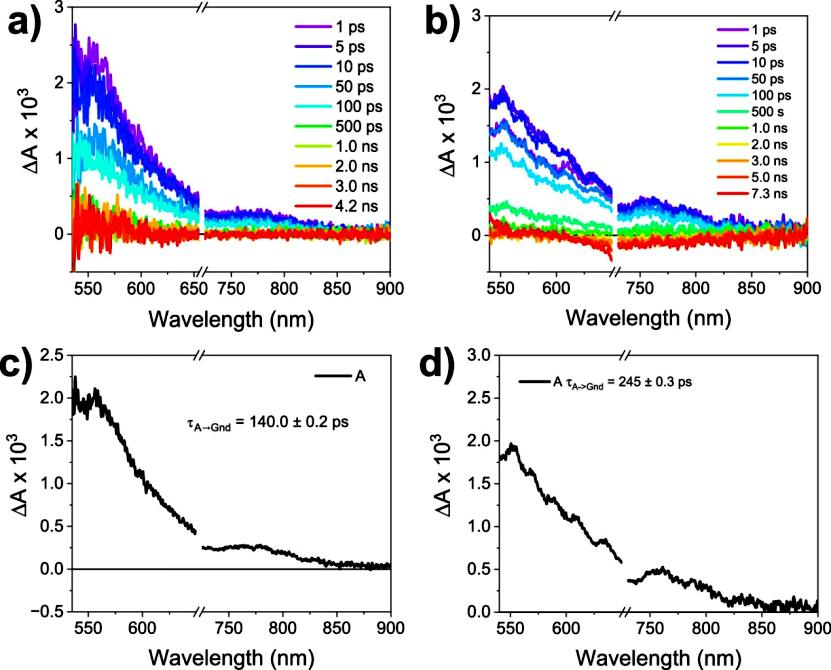
Figure 6. fs-TAM data for (−)-NDI-Δ-PER 1D and 2D. (a, b) fs-TAM spectra for (−)-NDI-Δ-PER 1D (λexc = 690 nm) and (−)-NDI-Δ-PER 2D (λexc = 690 nm) as a function of pump-probe delay; the probe light polarization direction for the 1D co-crystal is parallel to its long axis, while for the 2D co-crystal, it is aligned with the direction of maximum absorption. (c, d) Evolution-associated spectra for (−)-NDI-Δ-PER 1D using the A → GS model, and (−)-NDI-Δ-PER 2D using the SSA → GS, B → GS models.
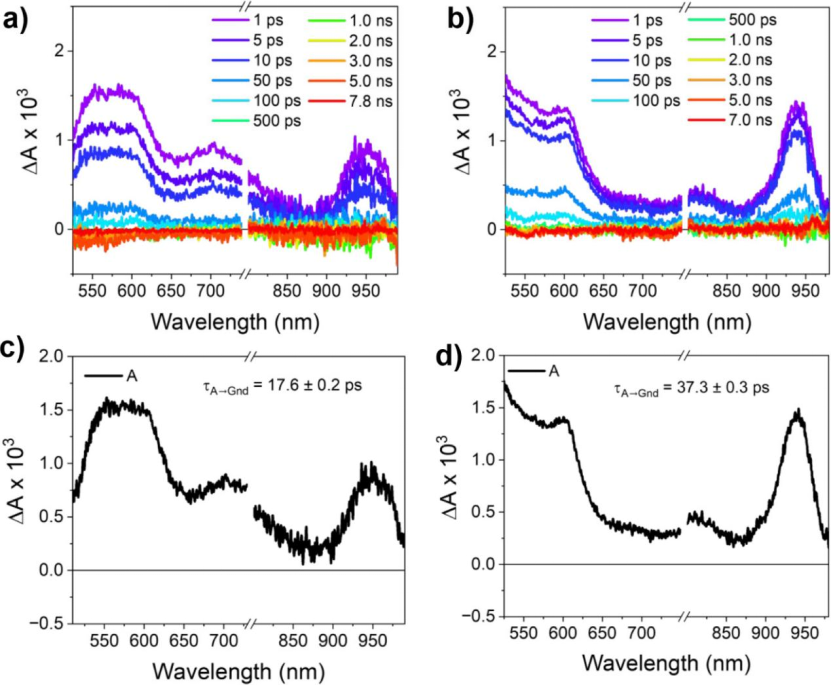
Figure 7. fs-TAM data for (−)-NDI-Δ-PXX 1D and 2D. (a, b) fs-TAM spectra for (−)-NDI-Δ-PXX 1D (λexc = 780 nm) and (−)-NDI-Δ-PXX 2D (λexc = 780 nm) as a function of pump-probe delay; the probe light polarization direction for the 1D co-crystal is parallel to its long axis, while for the 2D co-crystal, it is aligned with the direction of maximum absorption. (c, d) Evolution-associated spectra for both (−)-NDI-Δ-PXX 1D and 2D using the A → GS model.
4: Mechanism for Extending CT State Lifetime
As previously mentioned, in the (−)-NDI-Δ-PYR system, SOCT-ISC generates a long-lived 3CT state, a phenomenon similar to that observed in NDA-PYR co-crystals. In contrast, in the 1D and 2D co-crystals of (−)-NDI-Δ-PER and (−)-NDI-Δ-PXX, the photogenerated CT states rapidly recombine back to the ground state. Given that 1((−)-NDI•–-Δ-PER•+) has an energy of 1.7 eV, higher than the triplet energy of PER (1.53 eV), theoretically, its charge recombination process is expected to generate some 3*PER; however, the rapid decay of 1((−)-NDI•–-Δ-PER•+) to the ground state fully aligns with the theory of electron transfer rates: moderate CT state energy and strong coupling between the ground state and CT state lead to extremely fast recombination rates. Similar phenomena have been reported in other donor-acceptor systems. Therefore, the SOCT-ISC of 1((−)-NDI•–-Δ-PER•+) cannot compete dynamically with its decay to the ground state.
For the 1D and 2D co-crystals of (−)-NDI-Δ-PXX, the 1CT state energy is 1.6 eV, lower than the triplet energies of NDI (2.03 eV) and PXX (1.9 eV), making these triplet states energetically inaccessible (Figure 8). Again, it is emphasized that the lifetime of 1((−)-NDI•–-Δ-PXX•+) is shorter than that of 1((−)-NDI•–-Δ-PER•+), consistent with its energy being approximately 0.1 eV lower; according to the theory of electron transfer rates, this process lies in the Marcus inverted region of the rate-reaction free energy curve, predicting a faster recombination rate.
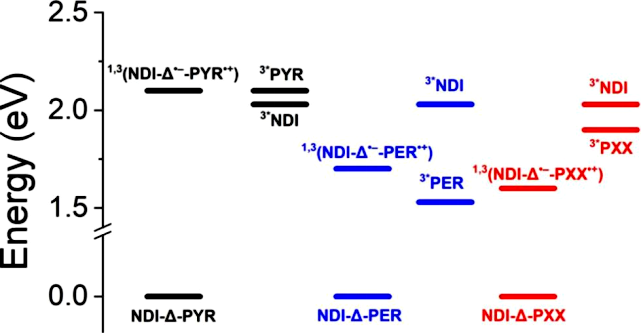
Figure 8. Energy level diagram of (−)-NDI-Δ-donor co-crystals. For the same donor-acceptor combination, the CT state energies of 1D and 2D co-crystals, as well as the energies of 1CT and 3CT, are roughly the same (in the figure, (−)-NDI-Δ is abbreviated as NDI-Δ).
In the needle-like 1D co-crystals of (−)-NDI-Δ-PYR, (−)-NDI-Δ-PER, and (−)-NDI-Δ-PXX, the lifetimes of the CT states are 1.1 ns, 140 ps, and 18 ps, respectively; while the corresponding lifetimes for the block-like 2D co-crystals are 2.4 ns, 245 ps, and 37 ps. The CT state lifetime in 2D co-crystals is consistently longer than in 1D co-crystals, attributed to the larger electronic coupling matrix elements for CT state charge recombination in the 1D crystals. We speculate that this change in electronic coupling directly arises from the structural changes induced by solvent guest molecules entering the (−)-NDI-Δ receptor units in the 1D co-crystals. This explanation is supported by time-dependent density functional theory (TD-DFT) calculations: directly extracting coordinates from the crystal structure, the contributions of the orbitals to the lowest CT transition and the oscillator strengths were calculated for (−)-NDI-Δ-donor. The results for the (−)-NDI-Δ-PYR co-crystal are shown in Figure 9.
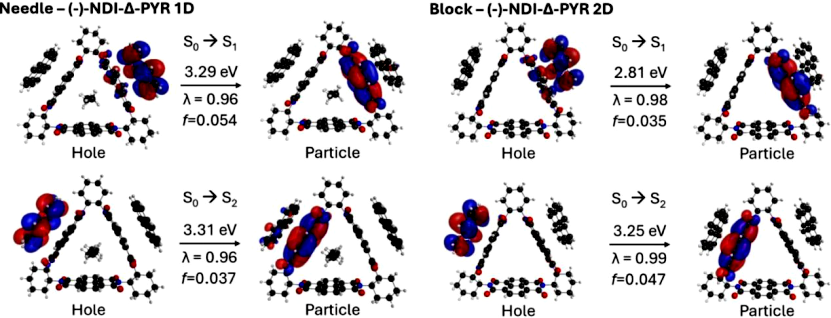
Figure 9. The lowest singlet electronic excitation, oscillator strength (f), λ value, and natural transition orbital (NTO) contributions for 1D and 2D (−)-NDI-Δ-PYR co-crystals. The λ value indicates the percentage contribution of the hole-particle pair in that transition.
TD-DFT calculation results show that in both 1D and 2D packed (−)-NDI-Δ-PYR co-crystals, the lowest energy CT transition is characterized by: the hole being fully localized on the PYR donor, while the electron (particle) is localized on an adjacent (−)-NDI-Δ receptor unit. Since these CT states can be directly excited by light, their lifetimes depend on the electronic coupling matrix elements, which can be directly associated with the transition oscillator strengths shown in Figure 9. The calculated oscillator strengths decrease from f = 0.054 for the (−)-NDI-Δ-PYR 1D co-crystal to f = 0.035 for the 2D co-crystal S0→S1 transition, fully consistent with the experimental observation that the CT state lifetime in the 2D co-crystal is approximately twice that of the 1D co-crystal. A similar comparison of the CT transition oscillator strengths for the (−)-NDI-Δ-PER co-crystal reveals that its S0→S1 and S0→S2 transitions are nearly degenerate in both 1D and 2D structures; thus, the significant contributions of the S0→S2 transition also lead to the same conclusion. Finally, the energy differences between the S0→S1 and S0→S2 transitions for the (−)-NDI-Δ-PXX co-crystal are quite large, again showing that the oscillator strength in the 2D structure is about half that of the 1D structure, corresponding to weakened electronic coupling, consistent with experimental observations.
Conclusion
This work employs a “solvent-dependent” strategy to successfully prepare a series of 1D and 2D donor-acceptor co-crystals. Using the NDI-based triangular molecule (−)-NDI-Δ as the acceptor and planar aromatic molecules PYR, PER, and PXX as donors, different dimensional single crystal co-crystals were self-assembled through donor-acceptor interactions, π-π stacking, solvent-π interactions, and hydrogen bonding non-covalent interactions. When the crystallization solvent (such as n-hexane or acetonitrile) can be encapsulated in the cavity of (−)-NDI-Δ, needle-like 1D single crystals are selectively grown; if a solvent that cannot be inserted into the cavity (such as dichloromethane or DMSO) is used, block-like 2D single crystals are obtained. Interestingly, due to the symmetry of (−)-NDI-Δ being disrupted in the 1D system while maintained in the 2D system, the CT exciton lifetime in the 2D co-crystal is significantly longer than that in the 1D co-crystal. This study demonstrates the design potential of “pre-organized covalent multipoint charge carriers” as co-crystal donors/acceptors, providing new insights for constructing advanced multifunctional materials with tunable CT characteristics.
DOI
10.1021/jacs.4c13800
Corresponding Author Profile

Michael R. Wasielewski: Professor of Chemistry at Northwestern University, Clare Hamilton Hall Chair, also an instructor in the Applied Physics program, and serves as the director of the Institute for Quantum Information Research and Engineering (INQUiRE) and the Center for Molecular Quantum Sensing (CMQT). Professor Wasielewski’s research focuses on photodriven processes in molecules and materials, artificial photosynthesis, molecular electronics, quantum information science, ultrafast spectroscopy, and time-resolved electron paramagnetic resonance spectroscopy. In recent years, he has published several articles as a corresponding author in top international journals, such as J. Am. Chem. Soc. 2024, 146, 27935-27945; Science, 2023, 382, 197-201; J. Am. Chem. Soc. 2023, 145, 14922-14931, etc.
▼For more exciting content, please long press the QR code▼ 
