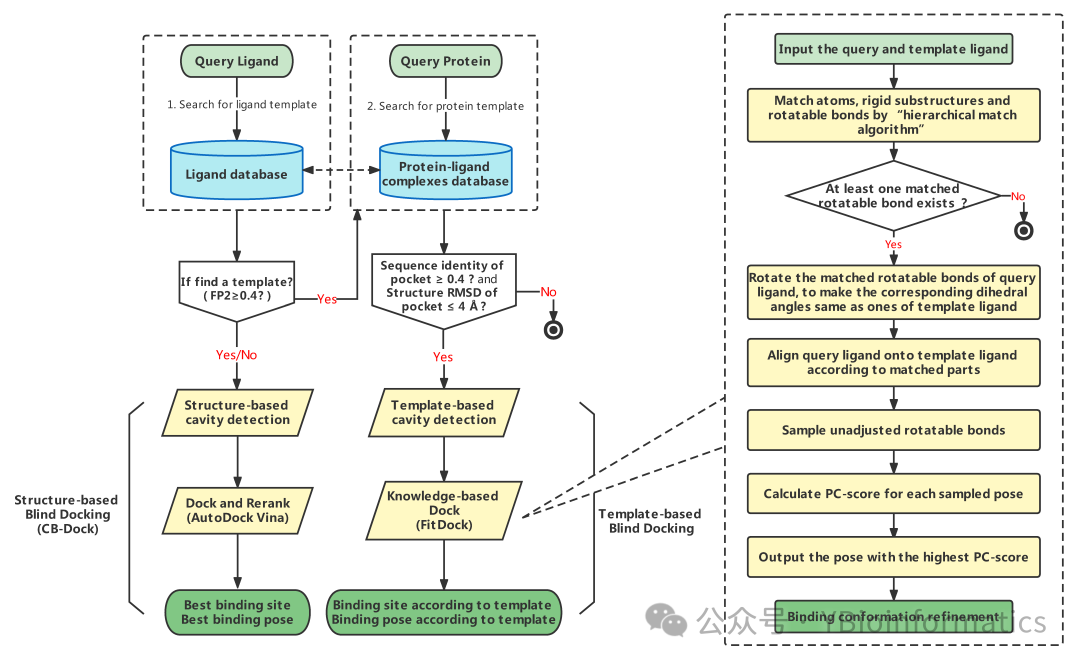
CB-Dock2 is an improved protein-ligand blind docking tool developed based on the original CB-Dock. Its workflow integrates three levels: traditional blind docking, homology template-assisted prediction, and fine-tuning optimization. First, it inherits the curvature-based pocket detection method from CB-Dock to identify surface pockets of the input protein and uses AutoDock Vina for initial blind docking, resulting in multiple potential binding sites and ligand conformations. On this basis, CB-Dock2 introduces a template-driven mechanism: the system retrieves template ligands from a built-in protein-ligand complex database that have a high topological similarity (FP2 ≥ 0.4) to the user-submitted ligand, and further calculates the sequence consistency between the target protein and these template proteins. If the similarity exceeds 40% and the RMSD of the template pocket is no more than 4 Å, the template and its binding site are retained for subsequent analysis. Utilizing this homology information allows for cavity detection and docking at more reasonable positions, thus avoiding potential biases in blind docking. Finally, CB-Dock2 refines the docking results using the built-in FitDock method: it first aligns the initial conformation of the ligand based on the template through multi-feature hierarchical alignment, and then explores various possible binding modes within this framework, optimizing for energy and geometry to output more reliable docking poses. Overall, CB-Dock2 significantly enhances the accuracy and reliability of binding site prediction and docking results while maintaining the flexibility of traditional blind docking by fully utilizing reference information from homologous complex pockets and combining it with FitDock’s conformation optimization.
 On the CB-Dock2 platform, to meet the needs of different users, two types of task workflows have been designed: “Search Cavities” and “Auto Blind Docking.” The former primarily focuses on pocket detection after inputting the protein and ligand, including cavity detection based on protein structure and template-assisted cavity detection in the presence of homologous templates; the latter automatically calls the above two types of programs, first detecting pockets and then performing molecular docking based on the detected binding sites.(1) Open the CB-Dock2 official website:
On the CB-Dock2 platform, to meet the needs of different users, two types of task workflows have been designed: “Search Cavities” and “Auto Blind Docking.” The former primarily focuses on pocket detection after inputting the protein and ligand, including cavity detection based on protein structure and template-assisted cavity detection in the presence of homologous templates; the latter automatically calls the above two types of programs, first detecting pockets and then performing molecular docking based on the detected binding sites.(1) Open the CB-Dock2 official website:
https://cadd.labshare.cn/cb-dock2/index.php
(2) Task Submission: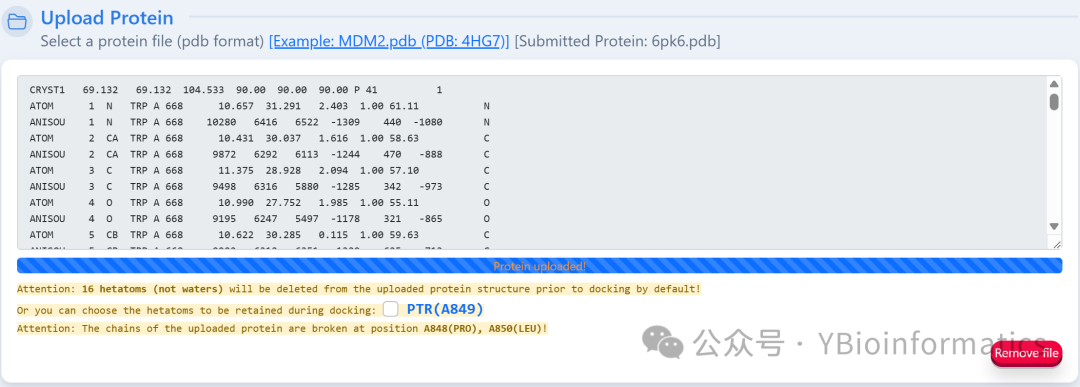 Users need to upload the PDB file of the target protein, and the system will automatically remove all heteroatoms (small molecules, ions, and water molecules), providing a warning message in the submission area to indicate whether heteroatoms are present. Users can choose to retain specific heteroatoms for docking. If the submitted protein is a biological assembly, it is recommended to split the chains first (Split Chains).
Users need to upload the PDB file of the target protein, and the system will automatically remove all heteroatoms (small molecules, ions, and water molecules), providing a warning message in the submission area to indicate whether heteroatoms are present. Users can choose to retain specific heteroatoms for docking. If the submitted protein is a biological assembly, it is recommended to split the chains first (Split Chains).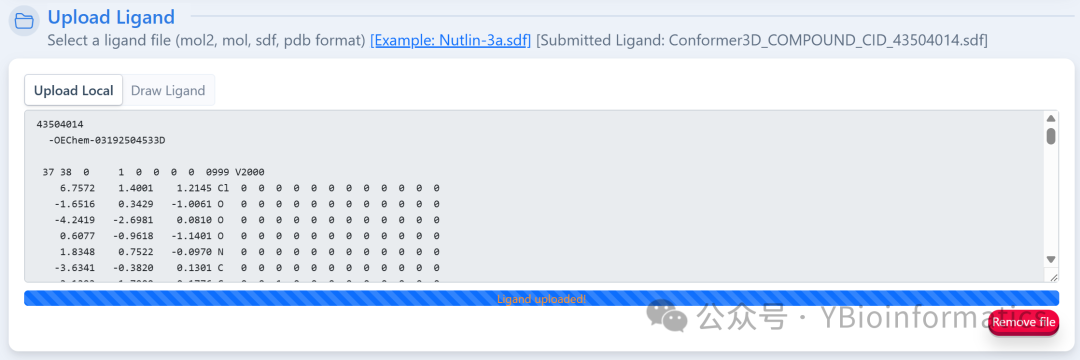 The ligand part can upload files in MOL2, MOL, SDF, or PDB format. If in MOL or SDF format, the system will convert it to PDB format using Open Babel; it can also be drawn online using the embedded JSME Molecule Editor.To ensure computational efficiency and the accuracy of AutoDock Vina, the ligand file size must be less than 15KB.
The ligand part can upload files in MOL2, MOL, SDF, or PDB format. If in MOL or SDF format, the system will convert it to PDB format using Open Babel; it can also be drawn online using the embedded JSME Molecule Editor.To ensure computational efficiency and the accuracy of AutoDock Vina, the ligand file size must be less than 15KB.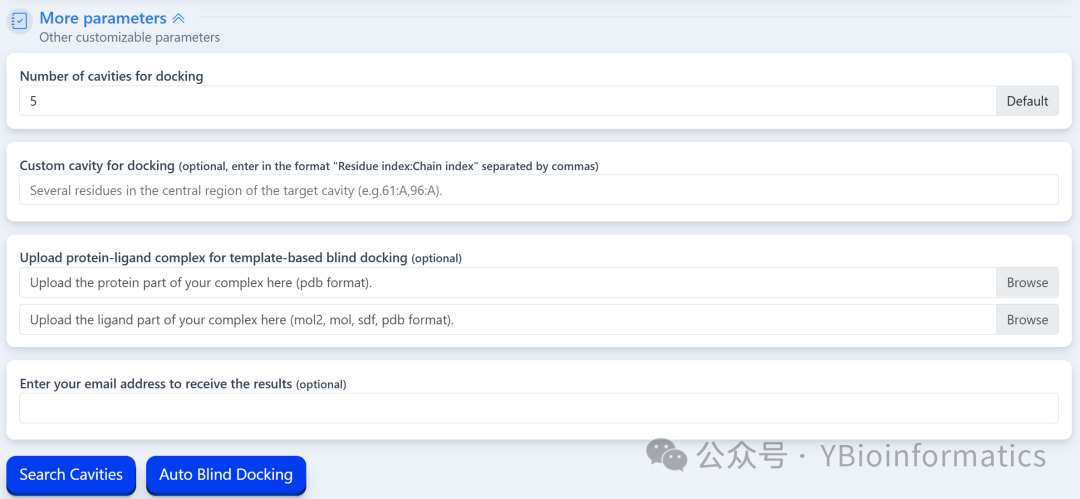 For the number of cavities, the system defaults to detecting 5 pockets using a non-template method. Users can customize this value through “More parameters,” but it is recommended not to exceed 20 to avoid prolonged run times and increased false positive rates. Additionally, users can choose to upload a protein-ligand complex as a template for template-driven blind docking; if the template is a biological assembly, the chains should also be split before uploading. Only one “Search Cavities” or “Auto Blind Docking” task can be submitted at a time. After the task is completed, users can click “View Result” in the task list to view or download results, or click “Remove” to delete results. If an email address is provided, the system will send the results via email. It is important to note that results from tasks not manually deleted will be automatically cleared after one day, so users should download and save output results in a timely manner.(3) Cavity Pocket Prediction Results:
For the number of cavities, the system defaults to detecting 5 pockets using a non-template method. Users can customize this value through “More parameters,” but it is recommended not to exceed 20 to avoid prolonged run times and increased false positive rates. Additionally, users can choose to upload a protein-ligand complex as a template for template-driven blind docking; if the template is a biological assembly, the chains should also be split before uploading. Only one “Search Cavities” or “Auto Blind Docking” task can be submitted at a time. After the task is completed, users can click “View Result” in the task list to view or download results, or click “Remove” to delete results. If an email address is provided, the system will send the results via email. It is important to note that results from tasks not manually deleted will be automatically cleared after one day, so users should download and save output results in a timely manner.(3) Cavity Pocket Prediction Results:
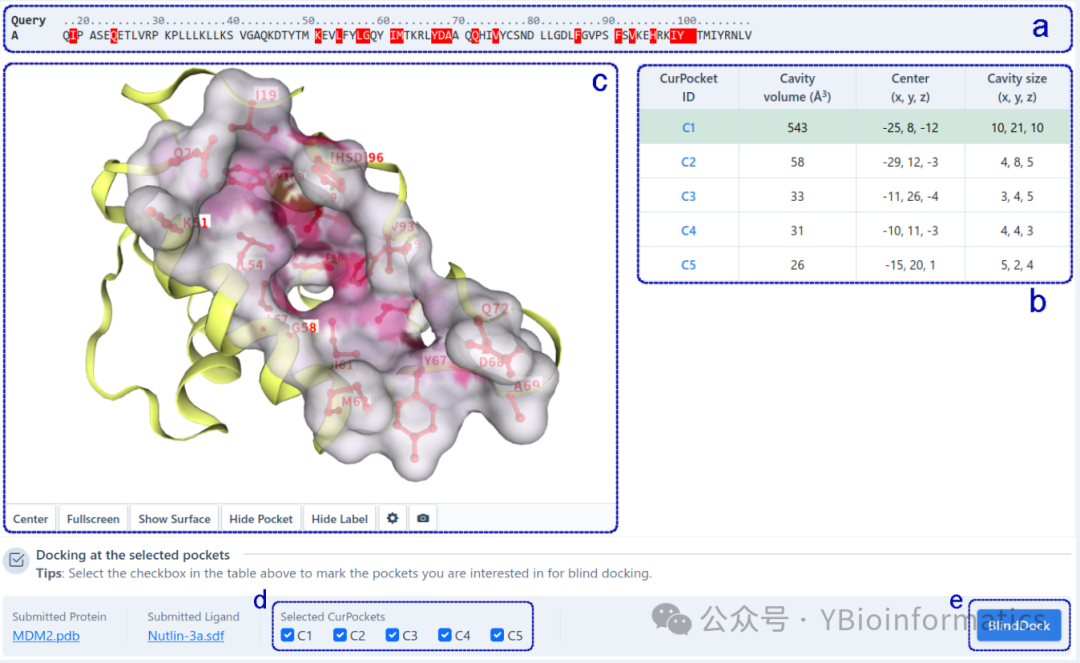
The cavity detection results interface is divided into two parts based on the detection method: structure-based cavity detection and template-based cavity detection (conducted in the presence of homologous templates). For structure-based cavity detection (as shown below), this part displays the complete sequence of the query protein (a) and highlights the residues in each detected cavity (b). Users can click on residues in the sequence list to display them in a 3D view (c). Detected cavities are sorted by volume, and users can click on different cavities in the table to view detailed structural information in 3D. Additionally, users can check cavities of interest (d) for molecular docking based on AutoDock Vina (e) to obtain potential binding modes of the query ligand in the target cavity.
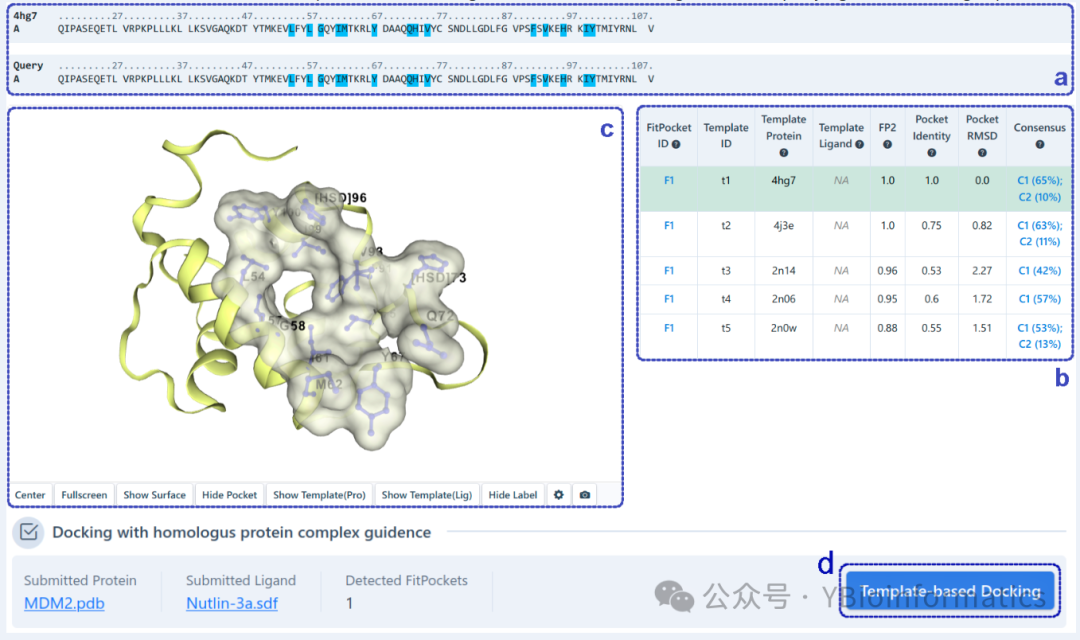
For template-based cavity detection (as shown below), this part displays the sequence alignment of the query protein with the template protein (a) and highlights the pocket residues detected based on the template (b). Similarly, users can click on residues in the sequence list to display them in a 3D view (c). Detected pockets are first sorted by FP2. If FP2 is the same, they are sorted by pocket identity. In the table (b), pockets detected from different templates but with the same “FitPocket ID” have at least 50% overlap in their pocket residues. Additionally, our server automatically provides a consistency analysis between pockets detected based on templates and those detected based on structure. Users can click on “Template-Library Docking” (d) to view the binding modes of the query ligand with the target pocket.
 (4) Blind Docking Prediction Results:
(4) Blind Docking Prediction Results:
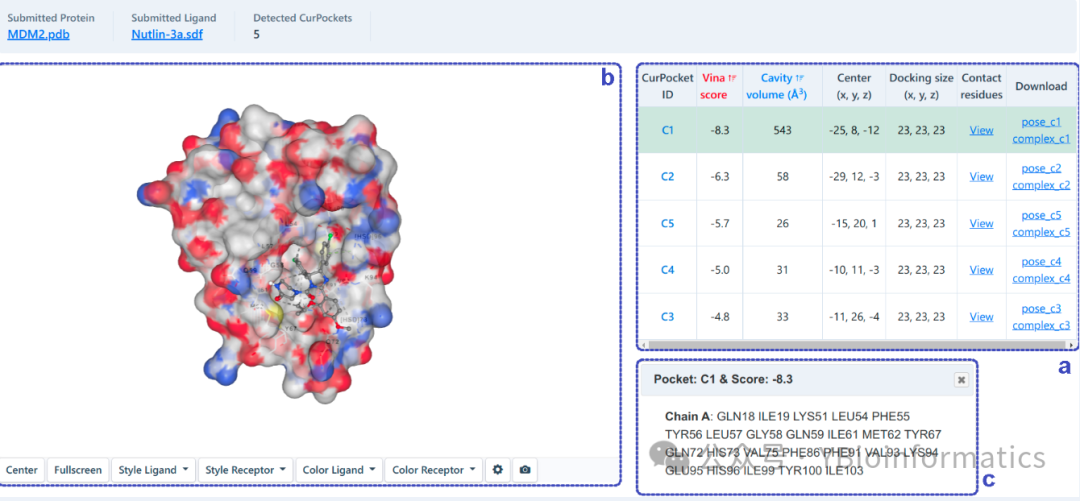
The results interface for automatic blind docking presents predictions based on two different blind docking principles. For structure-based blind docking (as shown below), we will perform docking based on AutoDock Vina in each detected cavity. We rank the potential binding sites of the query ligand based on the Vina score (kcal/mol). Users can click the “View” button in the table (a) to see the contact residues of the best binding mode in each cavity (c) along with structural information in the 3D view (b).
 Readers interested in services such as protein molecular docking, compound virtual screening, reverse docking, protein interaction prediction, molecular dynamics simulation, network pharmacology, lead compound optimization, and de novo protein design and modification can contact the author for communication and learning in the background~
Readers interested in services such as protein molecular docking, compound virtual screening, reverse docking, protein interaction prediction, molecular dynamics simulation, network pharmacology, lead compound optimization, and de novo protein design and modification can contact the author for communication and learning in the background~