
Authors: Wang Huijun, Lin Zhimiao
Affiliation: Dermatology Hospital, Southern Medical University, Guangzhou 510091, China
Corresponding Author: Wang Huijun, PhD, Assistant Researcher, E-mail: [email protected]
[Citation Format] Wang Huijun, Lin Zhimiao. Case report on skin fragility and woolly hair syndrome due to DSP gene mutation and literature review. Journal of Dermatology and Venereology, 2022, 29(3): 226-230.
[Funding Project] National Natural Science Foundation Youth Project (82003327)
Hereditary skin fragility diseases are a group of conditions characterized by skin and mucosal blistering or damage upon mechanical stress due to structural abnormalities, which may also involve hair, nails, and other systemic symptoms such as cardiac issues.[1] The latest classification standard from 2019 categorizes these diseases into several major types, including epidermolysis bullosa, epidermolytic keratolysis, erosive skin fragility diseases, and keratinization disorders with skin fragility, as well as connective tissue diseases with skin fragility.[1] These diseases are often caused by mutations in keratin or intercellular adhesion protein genes, leading to reduced protein quantity or structural disarray. This article reports a case characterized by increased skin fragility, hair abnormalities, and congenital pachyonychia, with the pathogenic gene identified as the DSP gene encoding desmoplakin through next-generation sequencing and Sanger verification, confirming the diagnosis of skin fragility-woolly hair syndrome. This study provides new evidence for exploring the correlation between DSP gene mutations and phenotypes.
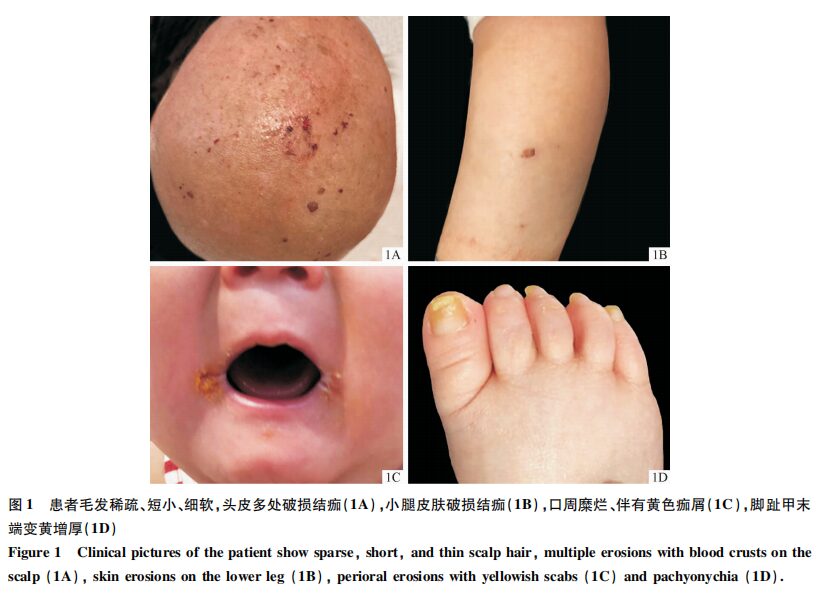
A 9-month-old male child presented with occasional blisters on the head, trunk, and limbs shortly after birth, with skin easily damaged upon friction. Multiple fingernails and toenails gradually turned yellow and thickened, with no significant hair growth observed. Dermatological examination revealed scattered ulcerations and crusting on the head and legs (Figure 1A, 1B), fine and sparse hair on the scalp, eyebrows, and eyelashes (Figure 1A), and fissures at the corners of the mouth with yellow crusts (Figure 1C). Mild desquamation of the toe skin and yellow thickening of several toenails were noted (Figure 1D). No significant palmoplantar keratoderma was observed. The patient’s height was 67 cm and weight was 7 kg, both below the normal value of -2SD. Cardiac ultrasound and electrocardiogram did not reveal any abnormalities. The parents were not closely related, and there was no similar history in the family.
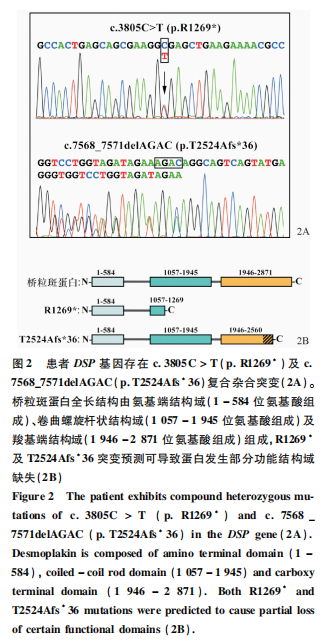
The average sequencing depth of the patient’s next-generation sequencing was 513.84, with 10× coverage at 99.26% and 20× coverage at 98.9%. The patient was found to carry suspicious pathogenic mutations in 19 genes, including DSP, MBTPS2, KRT16, TRPV3, FANCD2, TYRP1, GGCX, KRT4, HYAL1, SLC27A4, POFUT1, TRIM37, ANKRD11, etc. Family verification confirmed that only the compound heterozygous mutations in the DSP gene matched the family co-segregation, where c.3805C>T resulted in the 1,269th amino acid changing from arginine to a stop codon (p.R1269*), inherited from the father; the 7568_7571delAGAC mutation resulted in a frameshift (p.T2524Afs*36), inherited from the mother (Figure 2A). Based on the structural composition of desmoplakin, both mutations could lead to partial or complete loss of the C-terminal domain of the encoded desmoplakin (Figure 2B). Considering the patient’s clinical manifestations and genetic testing results, the diagnosis of skin fragility-woolly hair syndrome was made.
Desmosomes are critical structures for intercellular adhesion, expressed abundantly in tissues that endure mechanical stress, such as the epidermis and myocardium.[2] In the epidermis, desmosomes serve as bridges connecting the intermediate filament network of keratinocytes to the cell membrane.[3] Consequently, abnormalities in desmosomal structure not only affect intercellular adhesion but also reduce cytoskeletal stability, leading to increased skin fragility. Since the first discovery of the human desmosomal protein—mutations in the PKP1 gene encoding plakophilin 1 leading to ectodermal dysplasia skin fragility syndrome in 1997, various desmosomal proteins such as desmoplakin (encoded by the DSP gene), junctional plakoglobin (encoded by the JUP gene), desmoglein 1 and 4 (encoded by DSG1 and DSG4 genes), desmocollin 2 and 3 (encoded by DSC2 and DSC3 genes), and corneodesmosin (encoded by the CDSN gene) have been found to cause diseases affecting the skin, hair, or heart upon mutation, indicating the importance of adhesion proteins in maintaining the structural integrity and normal function of skin and its appendages.
Desmoplakin, encoded by the DSP gene, is the most abundant component of desmosomes, and alternative splicing results in two isoforms (DP-I and DP-II), with DP-I primarily expressed in the heart and DP-II mainly expressed in the skin.[6] Desmoplakin consists of a central rod domain and N-terminal and C-terminal domains, where the rod domain participates in dimerization, and the N-terminal domain controls its association with plakoglobin, playing a role in the organization of the cadherin-plakoglobin complex in desmosomes, while the C-terminal domain connects to the intracellular keratin intermediate filament network, playing a major role in adhesion between epidermal and muscle cells.[7]
Dominant missense mutations in the DSP gene often lead to arrhythmogenic cardiomyopathy, such as arrhythmogenic right ventricular dysplasia.[8-9] In rare cases, they may also cause severe dermatitis-multiple allergies-metabolic wasting syndrome.[10] In 1999, Armstrong et al. reported the first case of an autosomal dominant skin disease caused by insufficient haploinsufficiency of DSP mutations—striate palmoplantar keratoderma, which presented only with palmoplantar keratoderma without hair and other skin manifestations, possibly due to the higher mechanical stress in the palmoplantar regions requiring more and higher-quality desmosomal proteins to maintain their function, while skin in other areas could tolerate a reduction in half the amount. In 2000, a research team reported an autosomal recessive case of DSP mutation, where the patient presented with striate palmoplantar keratoderma along with woolly hair and left ventricular dilated cardiomyopathy, with the homozygous DSP gene truncation mutation leading to a lack of part of the C-terminal domain of the encoded desmoplakin.[12] Immunohistochemical staining of skin lesions from a patient carrying nonsense and missense compound heterozygous mutations revealed that the mutated desmoplakin localized to cell boundaries and also exhibited cytoplasmic localization. Electron microscopy showed acantholysis in the stratum corneum, reduced intercellular adhesion, and perinuclear condensation of keratin filaments, indicating that while the body can tolerate haploinsufficiency in some cases, the presence of other mutations can lead to more severe abnormalities in the intracellular keratin filament structure, causing more widespread and severe phenotypes. Literature reports summarize that the main phenotypes of recessive inheritance cases caused by DSP mutations include increased skin fragility, woolly hair or alopecia, palmoplantar keratoderma or keratinization in other areas, pachyonychia or nail dystrophy, and cardiac abnormalities, but due to varying ranges and severity, they have been classified based on clinical features into Carvajal syndrome, skin fragility-woolly hair syndrome, and lethal acantholytic epidermolysis bullosa, among others.[7,13-17] In this case, the patient presented with increased skin fragility, sparse and soft hair, and pachyonychia, and although no obvious woolly hair was observed, considering the patient is currently under 1 year old and hair has not fully developed, the possibility of developing woolly hair phenotypes in the future cannot be ruled out, thus the diagnosis remains as skin fragility-woolly hair syndrome.
The patient carried one nonsense mutation and one frameshift mutation, and although both mutations lead to premature translation termination, they occur at the C-terminal of the protein. Based on previously reported functional studies of nearby nonsense mutation sites, it is speculated that the two sites in this study likely have a lower probability of causing mRNA degradation mediated by nonsense mutations, leading to a higher possibility of producing truncated proteins. The c.3805C>T (p.R1269*) mutation would result in the formation of a protein lacking most of the coiled-coil rod domain and the entire C-terminal domain, while the p.T2524Afs*36 frameshift mutation from the mother would cause a change in the reading frame, resulting in 2,524 amino acids being altered and continuing to translate a small peptide structure composed of 36 other amino acids. Both mutations affect the C-terminal domain of the protein, which is primarily used for binding to the keratin intermediate filament network, suggesting a potential impact on the anchoring of keratin intermediate filaments to desmosomes, leading to instability in the cytoskeletal structure. Additionally, since the coiled-coil rod domain is involved in dimer formation, the p.R1269* mutation may also affect the dimer structure formation with the T2524Afs*36 mutant. The predicted structural alterations explain the reason for the increased skin fragility observed in the patient. The p.R1269* mutation site has been reported in a family with dominant inheritance of arrhythmias.[18] Currently, no cardiac abnormalities have been detected in this patient, but regular monitoring is necessary to prevent later cardiac changes.
Phenotypes caused by DSP mutations need to be distinguished from those caused by PKP1 gene mutations leading to ectodermal dysplasia/skin fragility syndrome. PKP1 encodes plakophilin 1, a major desmosomal assembly protein that can bind to desmoplakin and is similarly important for anchoring keratin intermediate filaments.[19] Patients carrying recessive mutations in PKP1 can present with skin fragility syndrome and ectodermal dysplasia affecting hair and nails, which overlaps significantly with the phenotypes caused by DSP recessive mutations, necessitating screening for pathogenic genes to confirm the diagnosis.
In summary, this study reports a case of skin fragility and woolly hair syndrome caused by DSP gene mutations, where the patient exhibited atypical features of increased skin fragility, hair abnormalities, and congenital pachyonychia. This case report expands the clinical phenotype spectrum caused by DSP gene mutations, aiding clinicians in improving disease diagnostic capabilities and providing new evidence for exploring the relationship between DSP gene mutations and phenotypes.
