
Click the blue text above to follow us
 Bisulfite Sequencing (BSP) is a DNA methylation detection technology based on bisulfite conversion. The core principle is to chemically treat DNA with bisulfite, converting unmethylated cytosine (C) to uracil (U), while methylated cytosine (5-methylcytosine, 5mC) remains unchanged. The converted DNA is amplified by PCR and sequenced, allowing the determination of the methylation status of each CpG site by comparing it to the original sequence.Key Reaction: Unmethylated C → U (appears as T during sequencing), Methylated C (5mC) → remains C.Detection Target: Methylation levels of CpG sites in specific genomic regions (such as promoters, enhancers, etc.).
Bisulfite Sequencing (BSP) is a DNA methylation detection technology based on bisulfite conversion. The core principle is to chemically treat DNA with bisulfite, converting unmethylated cytosine (C) to uracil (U), while methylated cytosine (5-methylcytosine, 5mC) remains unchanged. The converted DNA is amplified by PCR and sequenced, allowing the determination of the methylation status of each CpG site by comparing it to the original sequence.Key Reaction: Unmethylated C → U (appears as T during sequencing), Methylated C (5mC) → remains C.Detection Target: Methylation levels of CpG sites in specific genomic regions (such as promoters, enhancers, etc.).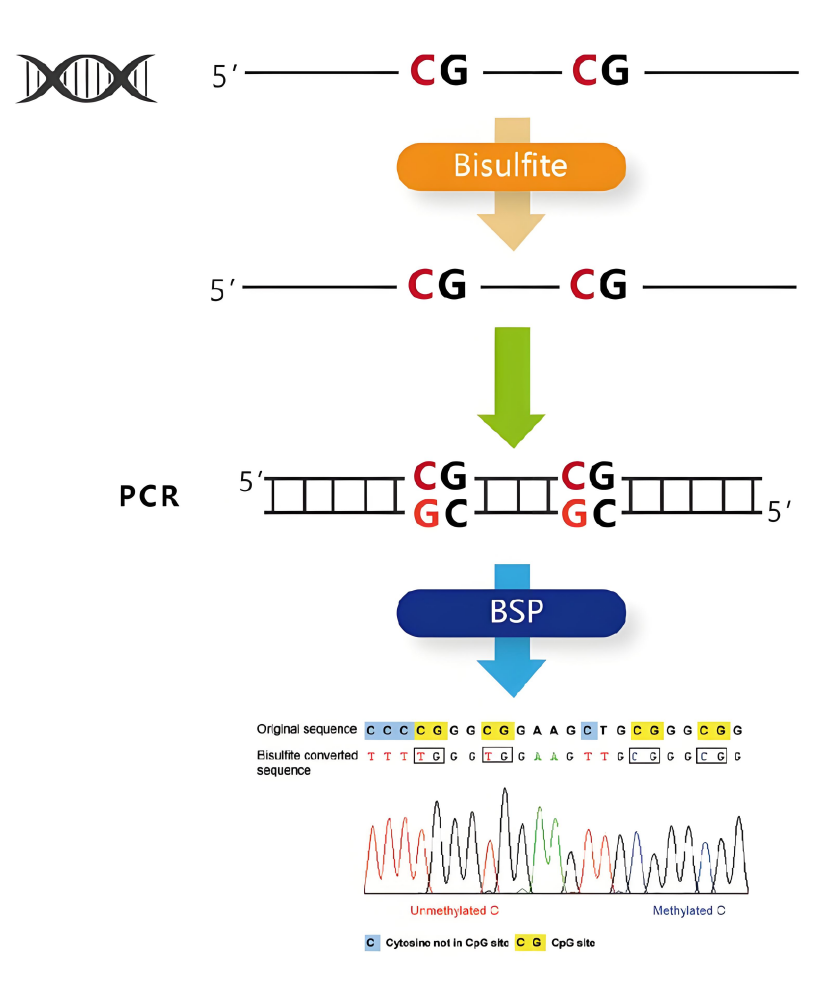
Technical Process
DNA Extraction and Quality Testing: Extract DNA from samples (such as cells, tissues, FFPE samples, etc.), ensuring DNA integrity (OD260/280≈1.8-2.0, no degradation).Bisulfite Conversion: Use a commercial kit for conversion, which includes the following steps: denature DNA into single strands; treat with bisulfite (usually heated under acidic conditions), deaminating unmethylated C to U; purify to remove bisulfite and modification products, resulting in unmethylated C converted to U and methylated C retained as C.Key Considerations: Conversion efficiency must be ≥99%, otherwise false positives may occur (unconverted unmethylated C may be misidentified as methylated).Primer Design and PCR Amplification
- Primer Design Principles: Primer sequences should not contain CpG sites (to avoid introducing methylation bias during amplification); design primers targeting the converted DNA sequence (U treated as T) to ensure specific binding to the target region; product length is typically 100-500 bp, facilitating subsequent cloning or direct sequencing.
- PCR Conditions: Use hot-start enzymes, low cycle numbers (to reduce non-specific amplification), and avoid methylation-dependent amplification bias (e.g., high GC content regions may have low amplification efficiency).
Sequencing and Methylation Analysis
- Direct Sequencing (Sanger Sequencing): Purify PCR products and directly sequence them, with each sequencing peak corresponding to a mixed methylation status of a sample (suitable for average methylation analysis of cell populations).
- Cloning Sequencing: Clone PCR products into vectors (such as pMD18-T), select single clones for sequencing, allowing analysis of the methylation patterns of individual DNA molecules (suitable for heterogeneous samples, such as tumor tissues).
- Data Analysis: Compare the original genomic sequence with sequencing results, calculate the C/T ratio for each CpG site, and determine the methylation frequency (methylation rate = C/(C+T) × 100%).
Technical Features
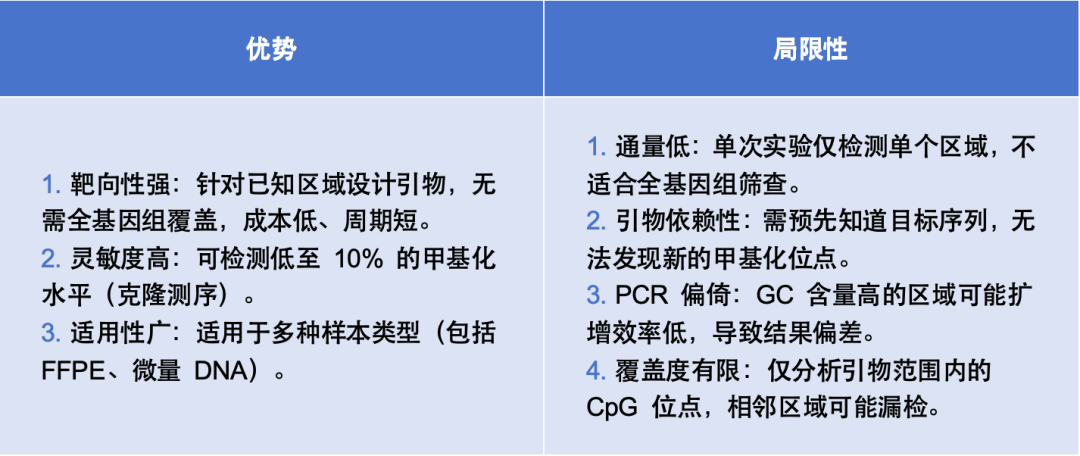
Main Application Areas
Epigenetics Research: Analyze the relationship between CpG methylation in gene promoters and enhancers and gene expression (e.g., high methylation of tumor suppressor gene promoters leading to transcriptional silencing).Cancer Research: Detect tumor-specific methylation markers (e.g., high frequency of APC and RASSF1A gene promoter methylation in lung and liver cancers); evaluate the therapeutic effects of DNA methylation inhibitors (e.g., 5-aza-dC).Disease Biomarker Screening: Analyze the methylation status of circulating free DNA in body fluids (such as blood and urine) for non-invasive detection (e.g., ctDNA methylation for early cancer diagnosis).Developmental Biology and Stem Cell Research: Track dynamic changes in methylation during cell differentiation (e.g., methylation remodeling during pluripotent stem cells differentiating into specific lineages).
Data Analysis Tools
Sequence Alignment: BSMAP, Bismark (suitable for high-throughput data); manual alignment (Sanger sequencing results).Methylation Quantification: QUMA (online tool), Methylation Analysis Suite (MAS, suitable for Illumina data), manual calculations in Excel (unit point analysis).Visualization: R language (ggplot2 for drawing methylation heatmaps), PyMeth (Python toolkit).
Comparison with Other Methylation Sequencing Technologies

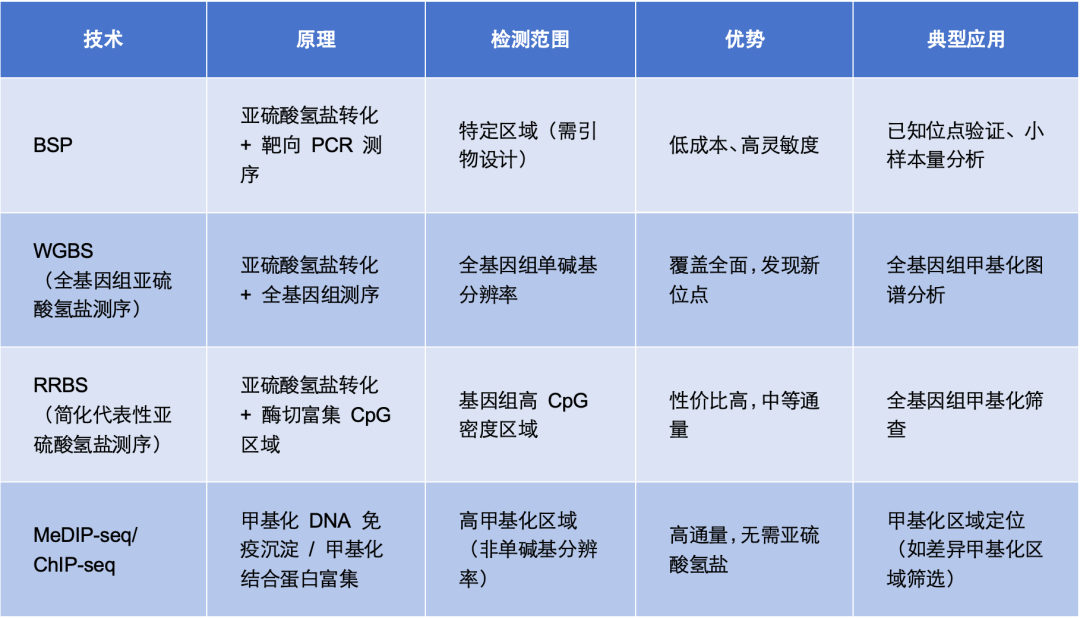

Considerations
Conversion Efficiency Validation: Must be assessed through positive/negative controls (e.g., known methylated/unmethylated DNA) to avoid false positive/negative results.Primer Design Tools: Common software includes Methyl Primer Express, MethPrimer, ensuring primers span multiple artificial SNPs (produced by C→T conversion) to avoid contamination of genomic DNA.Cloning Sequencing Quantity: If sample heterogeneity is high, increase the number of clones (typically 10-20 clones) to improve statistical reliability.Data Normalization: Avoid biases caused by differences in PCR cycle numbers and sequencing depth, it is recommended to use reference genes (such as ALU sequences) for calibration.
Previous Recommendations


 Reprint Instructions: Everyone is welcome to share this in WeChat Moments or groups; this article supports reprinting on the WeChat platform, thank you for your support!
Reprint Instructions: Everyone is welcome to share this in WeChat Moments or groups; this article supports reprinting on the WeChat platform, thank you for your support!