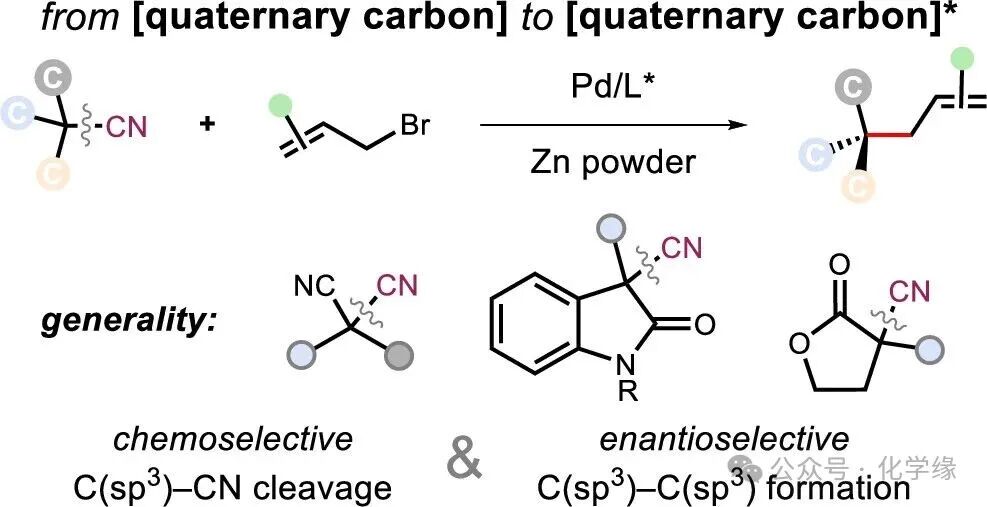
Professor Wenbo Liu’s team at Wuhan University proposed a unique asymmetric decyanative allylation (ADCNA) platform, utilizing tertiary nitriles as precursors to construct quaternary chiral centers through the cleavage of C-CN bonds and C(sp3)-C(sp3) cross-coupling. This strategy demonstrates high efficiency and enantioselectivity under mild conditions, benefiting from newly developed chiral ligands. The method has broad applicability, covering three classes of stable and readily available tertiary nitrile compounds: α,α-dialkyl malononitriles, α-cyano indolinones, and α-cyano lactones, thereby producing a diverse array of chiral-enriched linear and cyclic quaternary center compounds.
Abstract
Quaternary stereocenters are prevalent in natural products and bioactive molecules, where they influence the molecular structure, conformation, and function by enhancing the fraction of sp3 character. The enantioselective construction of such structures via cross-coupling remains a long-standing challenge, owing to the steric demands of forging C(sp3)-C(sp3) bonds and the limited accessibility of suitable tertiary carbon coupling partners. Here, we report a palladium-catalyzed asymmetric decyanative allylation (ADCNA) platform of C(sp3)-C(sp3) cross-coupling that transforms stable and readily available tertiary nitriles into enantioenriched acyclic and cyclic compounds bearing quaternary centers. This process proceeds via the selective addition of in situ-formed allyl zinc reagent to the cyano group, followed by retro-Thorpe-type C-CN bond cleavage and asymmetric allylation, guided by newly developed chiral ligands. The method exhibits a broad substrate scope across three classes of tertiary nitriles-malononitriles, α-cyano indolinones, and α-cyano lactones-offering high yields and excellent enantioselectivities under mild conditions. This research establishes a conceptually distinct retrosynthetic paradigm, from prochiral or racemic quaternary carbons to enantioenriched quaternary centers, enabled by chemoselective C-C bond cleavage and stereoselective C-C bond formation. The synthetic utility of this strategy is demonstrated by the downstream synthesis of a monoamine reuptake inhibitor and a CNGA2 channel blocker as well as synthetic intermediates of natural products physovenine and physostigmine.
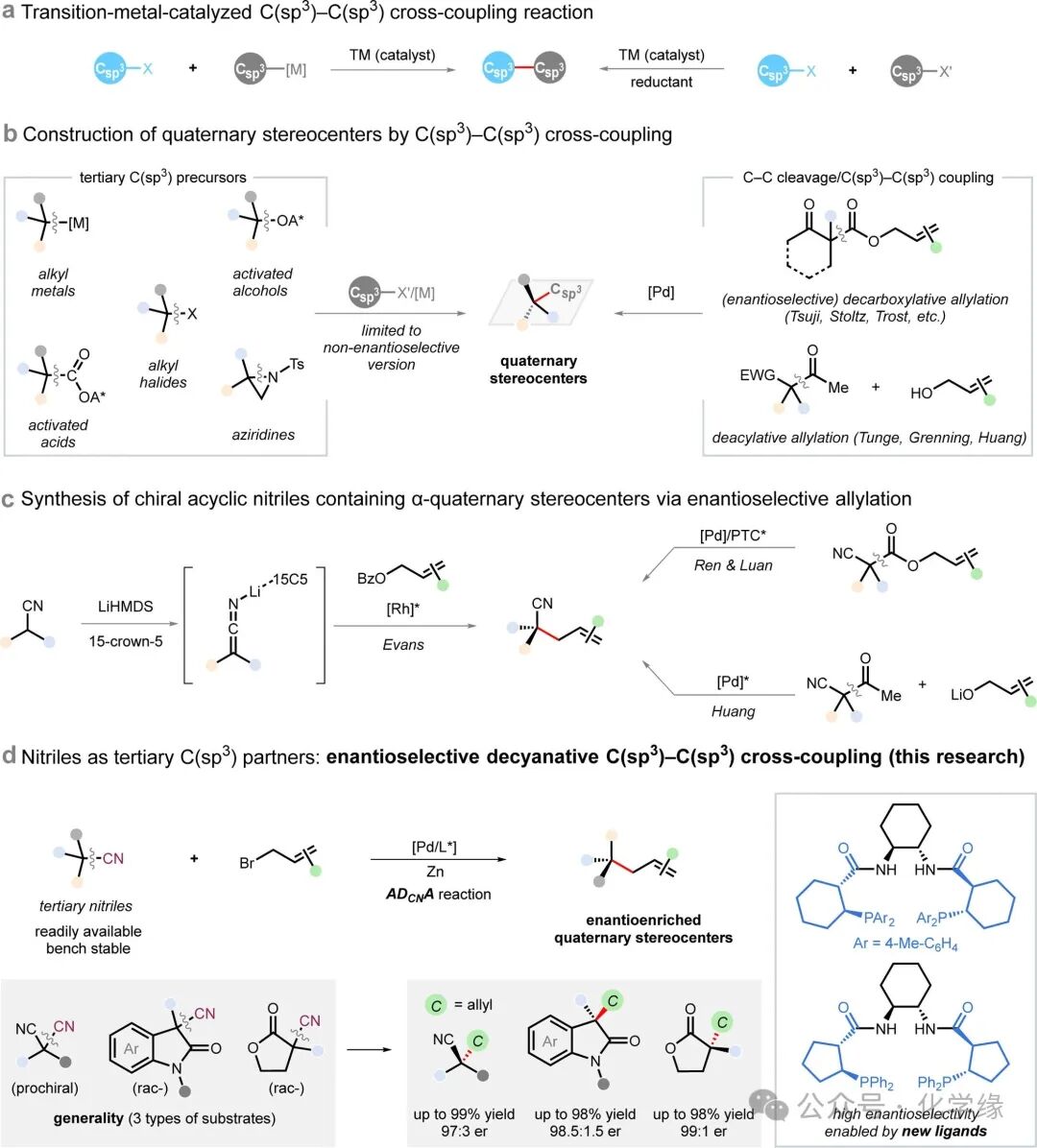
Figure 1 Overview of C(sp3)-C(sp3) cross-coupling reactions and their applications in the synthesis of quaternary carbon centers.
Transition metal-catalyzed cross-coupling reactions are one of the most significant breakthroughs in modern synthetic chemistry, fundamentally changing the way complex molecules are constructed. Traditional methods primarily focus on the formation of chemical bonds between C(sp2) substituents and carbon or heteroatom ligands, while recent breakthroughs in C(sp3)-C(sp3) cross-coupling technology have injected new vitality into classical fields, greatly expanding the accessible chemical space. However, constructing quaternary centers through cross-coupling—one of the most representative three-dimensional structural units prevalent in natural products and bioactive compounds—remains a persistent challenge. This difficulty arises from significant steric hindrance and competitive side reaction pathways such as β-hydride elimination, while the limited availability and inherent instability of tertiary halides and organometallic reagents further restrict their practical application value. Although methods utilizing activated tertiary alcohols, carboxylic acids, and nitrogen-containing compounds have emerged, these approaches generally lack control over enantioselectivity. Therefore, developing enantioselective cross-coupling strategies suitable for sterically congested C(sp3) substrates to construct quaternary centers remains an urgent issue in the field of transition metal catalysis.
Nitrile compounds are among the most versatile chemicals in synthetic chemistry, serving both as synthetic endpoints and as intermediates for downstream refinement. If the cyano group of prochiral or racemic nitrile compounds could be converted into other functional groups while achieving enantioselective construction of chiral centers, it would have profound value. However, the high bond dissociation energy of C-CN bonds hinders this transformation, and achieving control over enantioselectivity adds further obstacles. Existing methods primarily focus on the de-cyanation functionalization of aryl nitriles, while general strategies for the enantioselective de-cyanation transformation of tertiary nitriles have scarcely been reported.
The authors’ exploration stems from the successful development of chiral decarboxylative allylation reactions—a powerful tool for constructing quaternary chiral centers that has found widespread application in the synthetic field. In particular, the intramolecular decarboxylative allylation of allylic β-keto esters has become a benchmark strategy for achieving enantioselective C-C bond cleavage followed by C(sp3)-C(sp3) cross-coupling. Inspired by this, this paper proposes a unique asymmetric decyanative allylation (ADCNA) platform, utilizing tertiary nitriles as precursors to construct quaternary chiral centers through C-CN bond cleavage and C(sp3)-C(sp3) cross-coupling. This strategy demonstrates high efficiency and enantioselectivity under mild conditions, benefiting from newly developed chiral ligands. The method has broad applicability, covering three classes of stable and readily available tertiary nitrile compounds: α,α-dialkyl malononitriles, α-cyano indolinones, and α-cyano lactones, thereby producing a diverse array of chiral-enriched linear and cyclic quaternary center compounds.
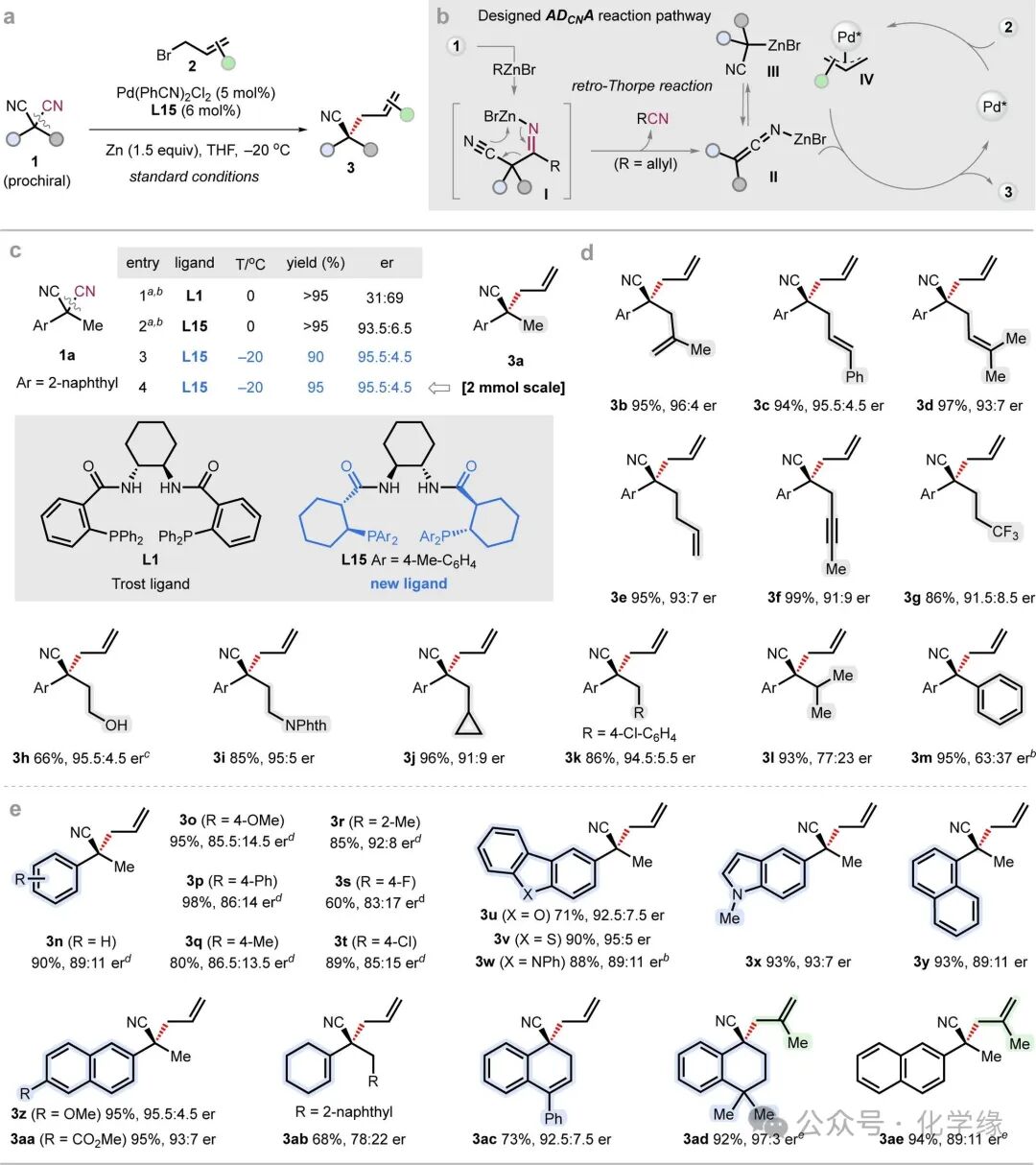
Figure 2 ADCNA reaction of malononitrile and its substrate scope.
Standard reaction conditions: 1 (0.2 mmol, 1 equiv), 2 (0.6 mmol, 3 equiv), Pd(PhCN)2Cl2(5 mol %), L15 (6 mol %), and Zn (0.3 mmol, 1.5 equiv) in THF (0.2 M) at -20 °C.
As shown in the mechanism depicted in Figure 2b, it is envisioned that the in situ-formed allyl zinc bromide adds to the cyano group of malononitrile 1, generating an imino zinc intermediate (I), which undergoes a retro-Thorpe-type cleavage reaction to yield the allyl imine species (II). This allyl imine can act as a nucleophile, undergoing a catalytic asymmetric coupling reaction with the allylic electrophile to produce enantiomerically enriched cyclic nitrile compounds with α-quaternary centers 3. Due to the challenges in controlling the geometric configuration of the allyl imine intermediate and the interconversion between N- and C-metalated forms (II and III), cyano anions have rarely been applied in catalytic enantioselective cross-coupling chemistry.
The developed asymmetric decyanative allylation reaction provides a conceptually distinct paradigm for chiral selective transition metal-catalyzed cross-coupling and quaternary center construction. Using malononitrile 1a as a model substrate, the exploration of this decyanative allylation reaction began. A series of ligands were examined in THF solvent at 0 °C. Using the catalyst generated in situ from Pd(MeCN)₂Cl₂ (5 mol %) and Trost ligand L1, the target allylation product 3a was obtained, but with low enantioselectivity (31:69 er). Screening of Trost-type ligands containing multiple stereocenters (L7–L10) revealed that using L10 significantly improved the enantioselectivity to 83.5:16.5. The absolute configuration of compound 3a is determined by the diamine backbone of the ligand. For example, L8 and L10 primarily generate the opposite configuration of enantiomer 3a, while L9 and L10 yield the same major enantiomer. Based on these findings, structural modifications of the ligand framework were pursued, leading to the synthesis of six new ligands (L11–L16). By expanding the ring size between the diphenylphosphine and amide from a six-membered ring to a seven-membered ring (L11) or reducing it to a five-membered ring (L12), enantioselectivity was found to decrease. Additionally, altering the diamine backbone (L13 and L14) did not enhance enantioselectivity, while introducing sterically more hindered backbones (L13) or substituents (L16) led to a significant drop in enantioselectivity.L15 ultimately emerged as the optimal ligand, not only providing excellent yields but also significantly enhancing enantioselectivity compared to L11.
To elucidate the enantioinduction process triggered by L15, a stereocontrol model was constructed based on DFT studies. This model indicates that the π-allyl group is deeply enveloped by the chiral pocket, while the 4-methyl on the phenyl substituent of the phosphine plays a crucial role in regulating enantioselectivity. In the unfavorable transition state (TS4-S), this methyl group experiences a repulsive interaction with the aryl substituent of substrate 1a, leading to a significant increase in energy barrier (compared to the favorable transition state TS4-R), with a ΔΔG‡ value of 2.5 kcal/mol. Further studies on reaction parameters indicate that under standard conditions at -20 °C, mixing Pd(PhCN)₂Cl₂ (5 mol%), L15 (6 mol%) with zinc powder (1.5 equivalents) in THF yields the product 3a with a 90% isolated yield and 95.5:4.5 enantioselectivity.
Subsequently, the substrate scope of malononitrile in this ADCNA reaction was evaluated. First, the substituents at the α-position of malononitrile were examined. Allyl substituents (3b–3d), homoallyl (3e), and propargyl (3f) were all compatible with the reaction, yielding quaternary chiral centers with enantioselectivities of 91:9–96:4 (er) and yields of 94-99%. Functional groups such as silyl ethers and ortho-phthalimides were also tolerated, yielding the corresponding products 3h and 3i with yields of 66% and 85%, respectively, and high enantioselectivity. Substrates containing fluorinated alkyl (3g), cyclopropyl methyl (3j), and 4-chlorobenzyl (3k) also achieved good yields with enantioselectivities of 91:9-94.5:5.5. In contrast, sterically bulky alkyl substituents (such as isopropyl and phenyl, 3l and 3m) still provided excellent yields but with significantly reduced enantioselectivity. These results indicate that the steric differences of the two substituents at the α-position of malononitrile are crucial. Subsequently, the applicability of aryl substituents was further explored. Various substituents on the phenyl ring were tolerated, yielding products 3n–3t with moderate to good yields and enantioselectivities. Fused aromatic frameworks including dibenzofuran (3u), dibenzothiophene (3v), carbazole (3w), indole (3x), and naphthalene derivatives (3y–3aa and 3ae) effectively participated in the reaction. Cyclohexenyl substituents (3ab) were also introduced, although with reduced enantioselectivity. Through this decyanative allylation strategy, cyclic quaternary ammonium stereocenters (3ac and 3ad) were successfully obtained.
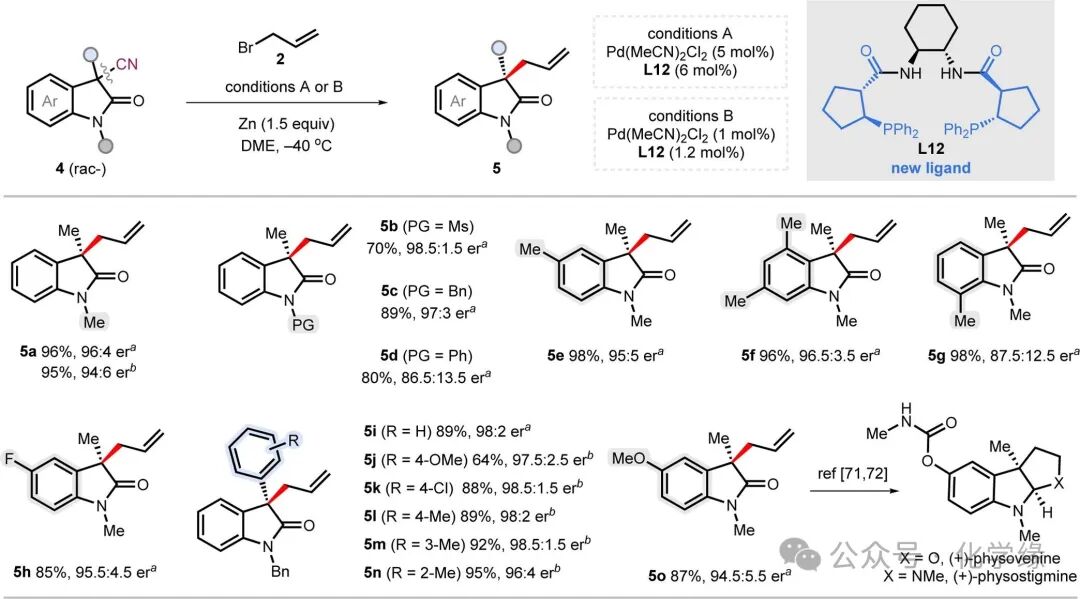
Figure 3 Substrate scope of indolinone compounds in ADCNA.
Standard reaction conditions: indolinone 4 (0.2 mmol, 1 equiv), allyl bromide 2 (0.6 mmol, 3 equiv), Pd(MeCN)2Cl2(5 mol % or 1 mol %), L12 (6 mol % or 1.2 mol %), and Zn (0.3 mmol, 1.5 equiv) in DME (0.05 M) at -40 °C.
The universality of the ADCNA strategy was further validated by other types of substrates. Indolinones, as a fundamental framework, are widely present in various alkaloid natural products. After rapid optimization of reaction conditions, the ADCNA scheme was confirmed to be applicable to indolinone substrates. Ligand L12 significantly outperformed L1 in terms of enantioselectivity (86:14 vs 70:30). By lowering the reaction temperature to -40 °C and switching to dimethyl ether as the solvent, further optimized conditions yielded product 5a with a yield of 96% and an enantioselectivity of 96:4. When the catalyst loading was reduced to 1 mol%, the enantioselectivity only slightly decreased (94:6). Investigation of different N-protecting groups on the indolinone framework showed that: methanesulfonyl (5b, 98.5:1.5 enantiomeric ratio) and benzyl (5c, 97:3 enantiomeric ratio) both maintained excellent results, while phenyl (5d, 86.5:13.5 enantiomeric ratio) led to a significant drop in enantioselectivity. Indolinones with different aryl substituents (including 5-methyl substituted 5e, 4,6-dimethyl substituted 5f and 5-fluoro substituted 5h) all achieved yields of 85-98% and enantioselectivities of 95:5-96.5:3.5. In contrast, 7-methyl substituted indolinone (5g) resulted in decreased enantioselectivity (87.5:12.5 er). When the α-substituents of indolinone shifted from alkyl to aryl (5i–5n), the reaction proceeded efficiently, providing excellent yields and enantioselectivities (96:4-98.5:1.5 er). As key intermediates in the total synthesis of physovenine and physostigmine, the 5-methoxy pyridinone 5o was obtained with a yield of 87% and an enantioselectivity of 94.5:5.5.
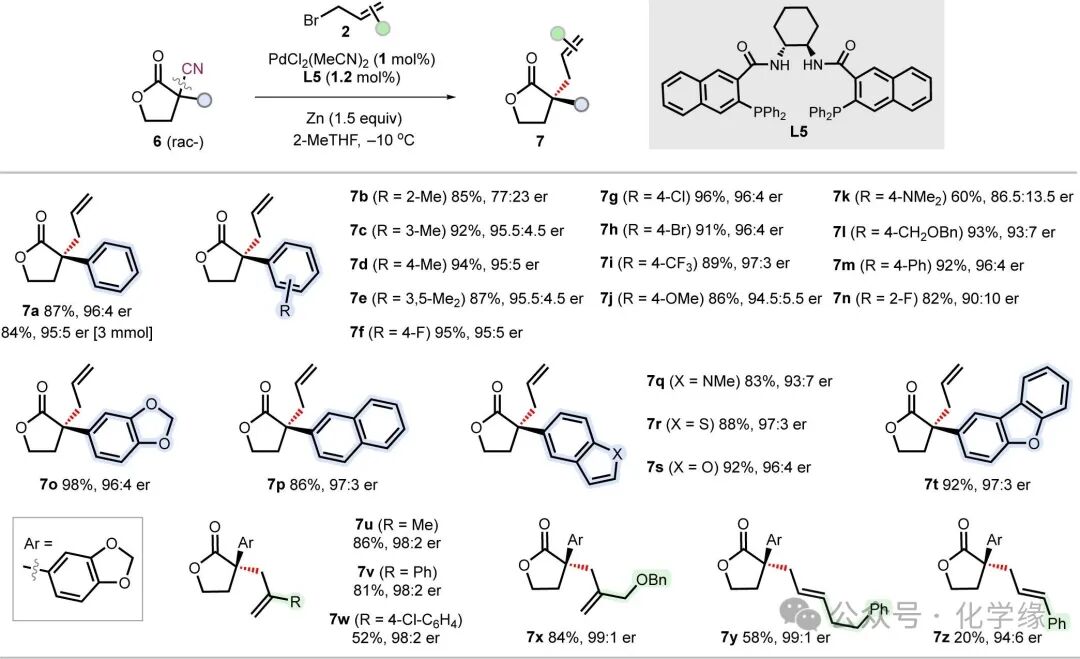
Figure 4 Substrate scope of lactone compounds in ADCNA.
Standard reaction conditions: lactone 6 (0.2 mmol, 1 equiv), allyl bromide 2 (0.6 mmol, 3 equiv), Pd(MeCN)2Cl2(1 mol %), L5 (1.2 mol %), and Zn (0.3 mmol, 1.5 equiv) in 2-MeTHF (0.1 M) at -10 °C.
Given the value of lactones in synthesis and their underutilization in enantioselective allylation reactions, the authors evaluated the ADCNA reaction using α-cyano lactone 6 as a substrate. Rapid ligand screening using model substrate 6a identified L5 as the optimal ligand, enabling the product 7a to be generated with a yield of 87% and an enantiomeric ratio of 96:4, achieving a balance between structural simplicity and performance, comparable to L15. The reaction exhibited high reactivity and selectivity, requiring only 1 mol% of palladium loading, and maintained yield and enantioselectivity even when scaled up to 3 mmol .
Substrates with meta- and para-substituted aryl groups (7c–7m) and differing electronic properties (such as methyl, methoxy, halogens, trifluoromethyl) had minimal impact on enantioselectivity, except for dimethylamino substituents (7k). The enantioselectivity of ortho-substituted aryl groups (7b and 7n) significantly decreased, which correlates with increased steric hindrance. Fused aromatic substituents (including benzodioxole-5-yl (7o), naphthyl (7p), indolyl (7q), benzothiophenyl (7r), benzofuranyl (7s), and dibenzofuranyl (7t) were all well tolerated in the reaction, ultimately yielding 83-98% yields and 93:7-97:3 enantioselectivities. Subsequently, the substrate scope of allylic electrophiles was further expanded by reacting with lactone 6o, discovering that 2-substituted allyl bromides (7u–7x) were not only viable but exhibited higher enantioselectivity compared to unsubstituted allyl bromides. The use of 3-alkyl and 3-phenyl allyl bromides (7y and 7z) resulted in lower yields due to increased steric hindrance, yet still achieved high enantioselectivities (94:6-99:1 er). Although α-benzyl substituted γ-lactones (6u were not feasible in the palladium-catalyzed system, an alternative nickel-catalyzed system still yielded products 7u with moderate yields and enantioselectivity. Finally, the reactivity of α-cyano δ-lactone substrates was examined. Unlike γ-lactones, no decyanative allylation occurred, but rather a decarboxylative allylation pathway predominated, generating nitrile compounds containing useful linear quaternary ammonium stereocenters with moderate enantioselectivity.
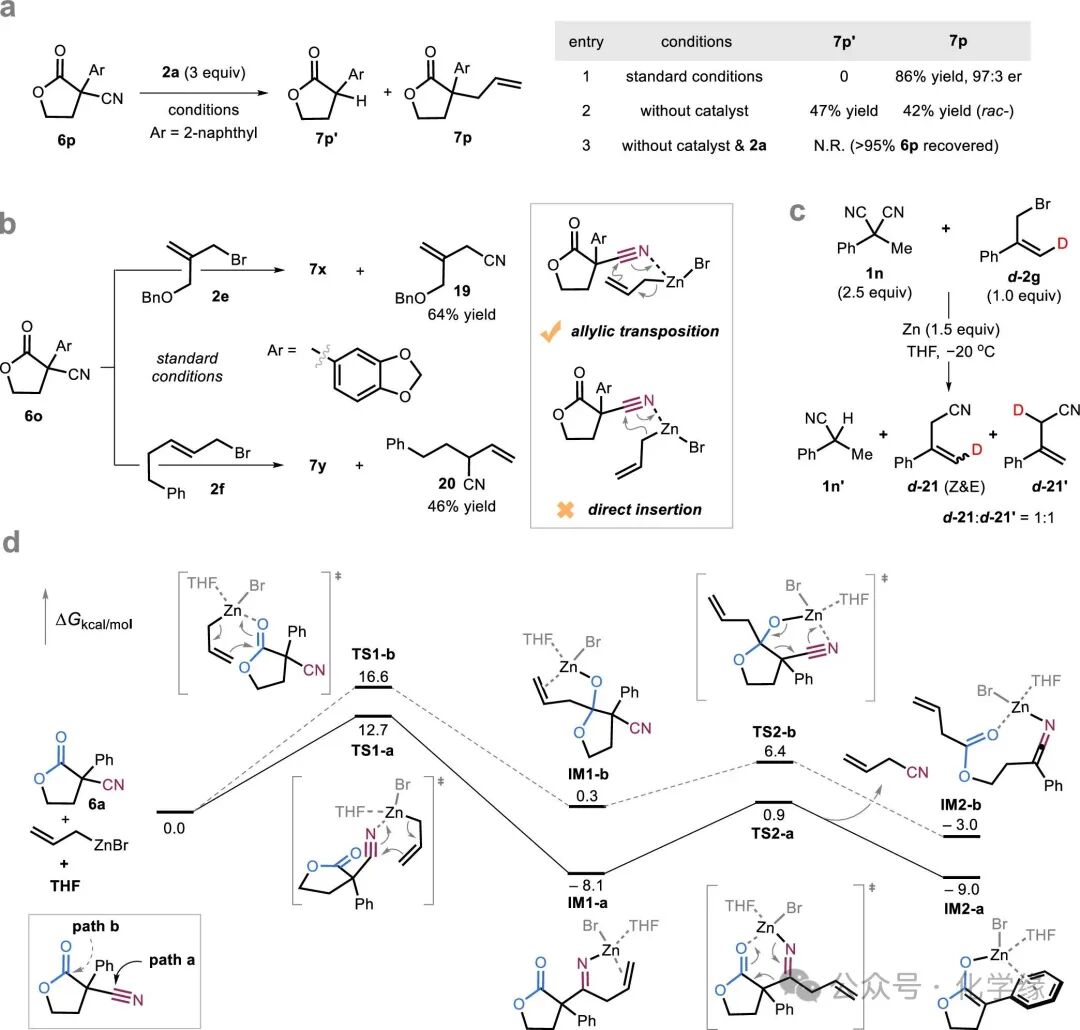
Figure 5 Mechanistic studies.
To gain deeper insight into the mechanism of the ADCNA reaction, particularly when unstable lactones are used as substrates, a series of control experiments were conducted. In the absence of a palladium catalyst, 6p reacted to simultaneously generate de-cyanated protonated products 7p′ (47% yield) and racemic allylation products 7p (42% yield), indicating that the zinc/brominated allyl system can efficiently and selectively cleave C-CN bonds but is prone to strong background reactions. Conversely, when zinc powder was used in the absence of allyl bromide 2a, no reaction occurred, and the starting material 6p was completely recovered. The high enantioselectivity observed under standard reaction conditions indicates that the palladium catalyst significantly accelerates the reaction rate.
DFT calculations indicate that pathway a describes the addition of allyl zinc to the cyano group, followed by retro-Thorpe-type C-C bond cleavage, eliminating the cyano group to generate an ester enol. In contrast, pathway b represents a retro-Claisen process, releasing the ester moiety. Calculations show that the energy barriers for the addition step (TS1-a) and subsequent C-C bond cleavage (TS2-a) in pathway a are significantly lower than those of the competing retro-Claisen pathway (TS1-b and TS2-b). Consistent with this, intermediates IM1-a and IM2-a are more stable than their corresponding IM1-b and IM2-b. The enhanced stability of IM2-a is attributed to favorable zinc-π interactions between the magnesium atom in the zinc enol salt and the phenyl substituent, making it thermodynamically more favorable than the zinc-keto imine intermediate formed through acyl cleavage, IM2-b.
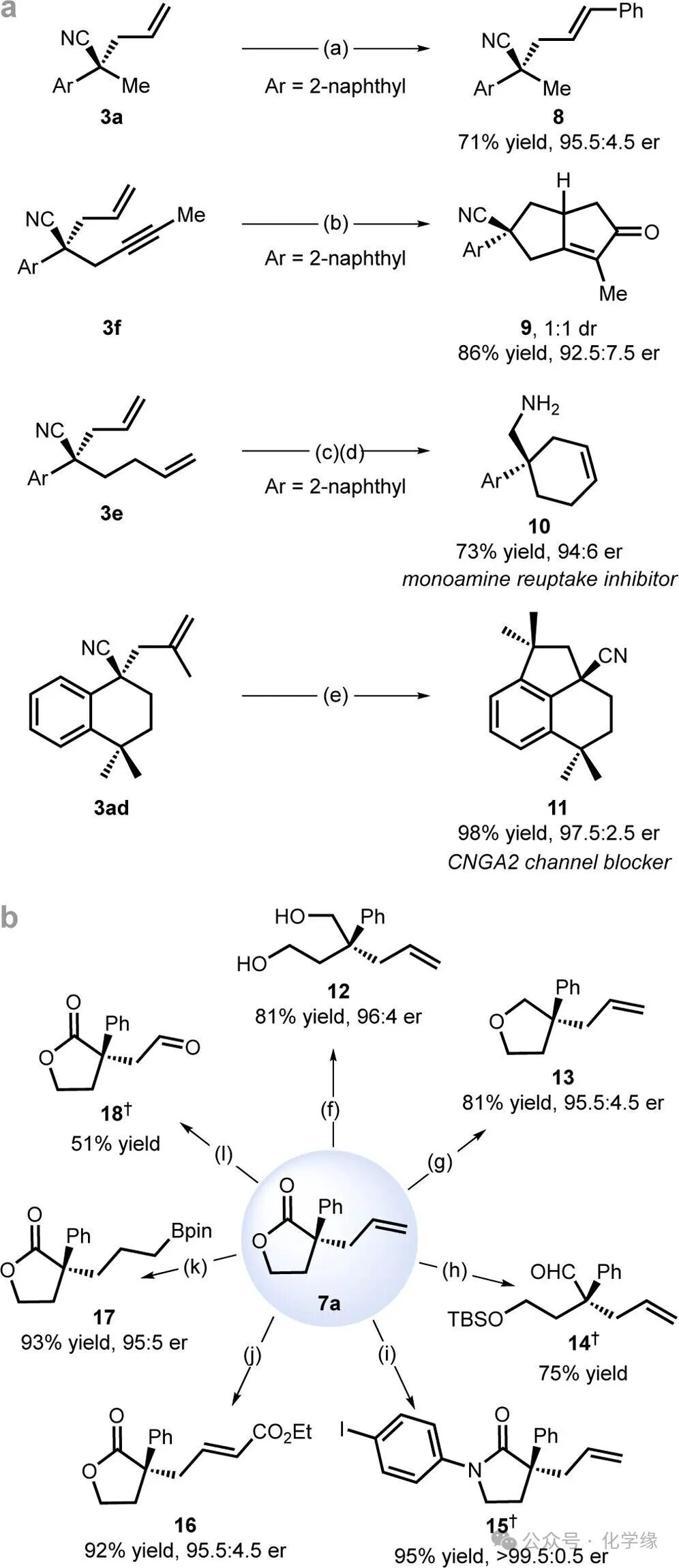
Figure 6 Synthetic applications.
The synthetic practicality of this method is demonstrated. Nitrile products containing a cinnamaldehyde group 8 can be obtained through olefin metathesis with styrene. The 1,6-diyne 3f underwent a Pauson–Khand reaction to achieve rapid construction of bicyclic [3.3.0] octane 9. The monoamine reuptake inhibitor 10 can be directly obtained from 3e through olefin metathesis, followed by nitrile reduction. Meanwhile, the cyclic product 3ad underwent a one-step Friedel–Crafts alkylation reaction to achieve the enantioselective synthesis of the tricyclic compound 11, which is a blocker of the cyclic nucleotide-gated ion channel (CNGA2). Depending on the reaction conditions, lactone 7a can be reduced to yield diols 12, tetrahydrofuran 13 and aldehyde 14. The lactam 15, which has synthetic value, can be efficiently prepared in a one-step reaction, providing a concise synthetic route for lactams containing α-quaternary centers.
Conclusion: A general and efficient asymmetric decyanative allylation (ADCNA) strategy has been developed, achieving chemoselective C-CN bond cleavage/enantioselective C(sp3)-C(sp3) cross-coupling, transforming tertiary nitriles and allyl bromides into quaternary chiral centers. This method encompasses three different classes of nitrile compounds, featuring advantages such as low catalyst loading, broad applicability, strong functional group compatibility, and scalability. The newly developed chiral ligands play a crucial role in achieving high enantioselectivity, and the resulting enantiomerically enriched products containing quaternary centers serve as multifunctional intermediates, capable of undergoing a series of downstream transformations to generate valuable building blocks, bioactive molecules, and key precursors of natural products, fully demonstrating the synthetic practicality of this method.
Article Information:
Catalytic Enantioselective C(sp3)–C(sp3) Cross-Coupling of Tertiary Nitriles with Allyl Halides to Quaternary Stereocenters
Gui-Feng Ding, Zi-Hao Chen, Jiang-Lian Deng, Yu-Qing Zheng, Xu-Dong Hu, and Wen-Bo Liu*
DOI: 10.1021/jacs.5c15414
We hope this article inspires you, and let us work together for a better future!
